Chronische Eosinophilenleukämie (CEL)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:obligat
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
Auf Basis der aktuellen Leitlinien und des aktuellen Forschungsstandes ergeben sich verschiedene diagnostische Empfehlungen für Patienten mit chronischer Eosinophilenleukämie (CEL). Wir haben Ihnen die wichtigsten Infos zur Klassifikation und den diagnostischen Methoden am MLL zusammengefasst. Zudem haben wir weiterführende Links und Literatur zur Prognose und Therapie bei CEL zusammengestellt, damit Sie sich tiefergehend informieren können.
Chronische Eosinophilenleukämie: Klassifikation
Die chronische Eosinophilenleukämie (CEL) ist durch eine klonale Eosinophilie charakterisiert und wird den myeloproliferativen Neoplasien (MPN) zugeordnet. Es ist eine sehr seltene und aggressive Erkrankung, bei der die klonale Proliferation eosinophiler Vorläuferzellen eine Erhöhung der Eosinophilen im peripheren Blut, Knochenmark und peripheren Gewebe bewirkt. Eine leukämische Infiltration oder die Freisetzung von Zytokinen, Enzymen oder anderen Proteinen der Eosinophilen können zu Organschäden von z.B. Herz, Lunge, Haut, Gastrointestinaltrakt oder dem zentralen Nervensystem führen (WHO 2022).
Die Diagnose einer CEL wird meistens durch den Ausschluss anderer hämatologischer Neoplasien, die eine Eosinophilie aufweisen können, gestellt. Folgende Kriterien gelten gemäß WHO 2022 zur Diagnosestellung (WHO 2022):
- Eosinophilie (Eosinophilenzahl >1,5 x 109/L) im peripheren Blut bei mindestens zwei Untersuchungen über ein Intervall von mindestens 4 Wochen
- Die WHO-Kriterien für MPN, MDS/MPN, MLN-eo, Mastozytose oder AML sind nicht erfüllt
- Es liegt kein PDGFRA-, PDGFRB- oder FGFR1-Rearrangement vor, kein ETV6::ABL1-Rearrangement sowie keine JAK2- und FLT3-Rearrangements
- Es liegt eine klonale zyto- oder molekulargenetische Veränderung vor (die Möglichkeit einer klonalen Hämatopoese unbestimmten Potentials (CHIP) sollte in Betracht gezogen werden)
- Es liegt eine aberrante Morphologie im Knochenmark (z.B. Megakaryozyten oder erythroide Dysplasie)
Lässt sich keine Klonalität durch den Nachweis einer zyto- oder molekulargenetischen Veränderung nachweisen, ist die Diagnose eines idiopathischen hypereosinophilen Syndroms (HES) zu stellen. Dieses zeichnet sich durch eine für ≥4 Wochen andauernde Eosinophilenzahl von ≥1,5 x 109/L aus, für die keine zugrundeliegende Ursache nachgewiesen werden kann. Zudem liegen Anzeichen für die Involvierung von Organen und deren Dysfunktion vor. Ohne diese Anzeichen sollte die Bezeichnung idiopathische Hypereosinophilie verwendet werden. Eine Eosinophilie kann auch durch die vermehrte Freisetzung von T-Zell-assoziierten Zytokinen bewirkt werden. Daher sollte das Vorliegen aberranter T-Zellen ausgeschlossen werden. Einen Überblick zur Differentialdiagnostik gibt Abbildung 1.
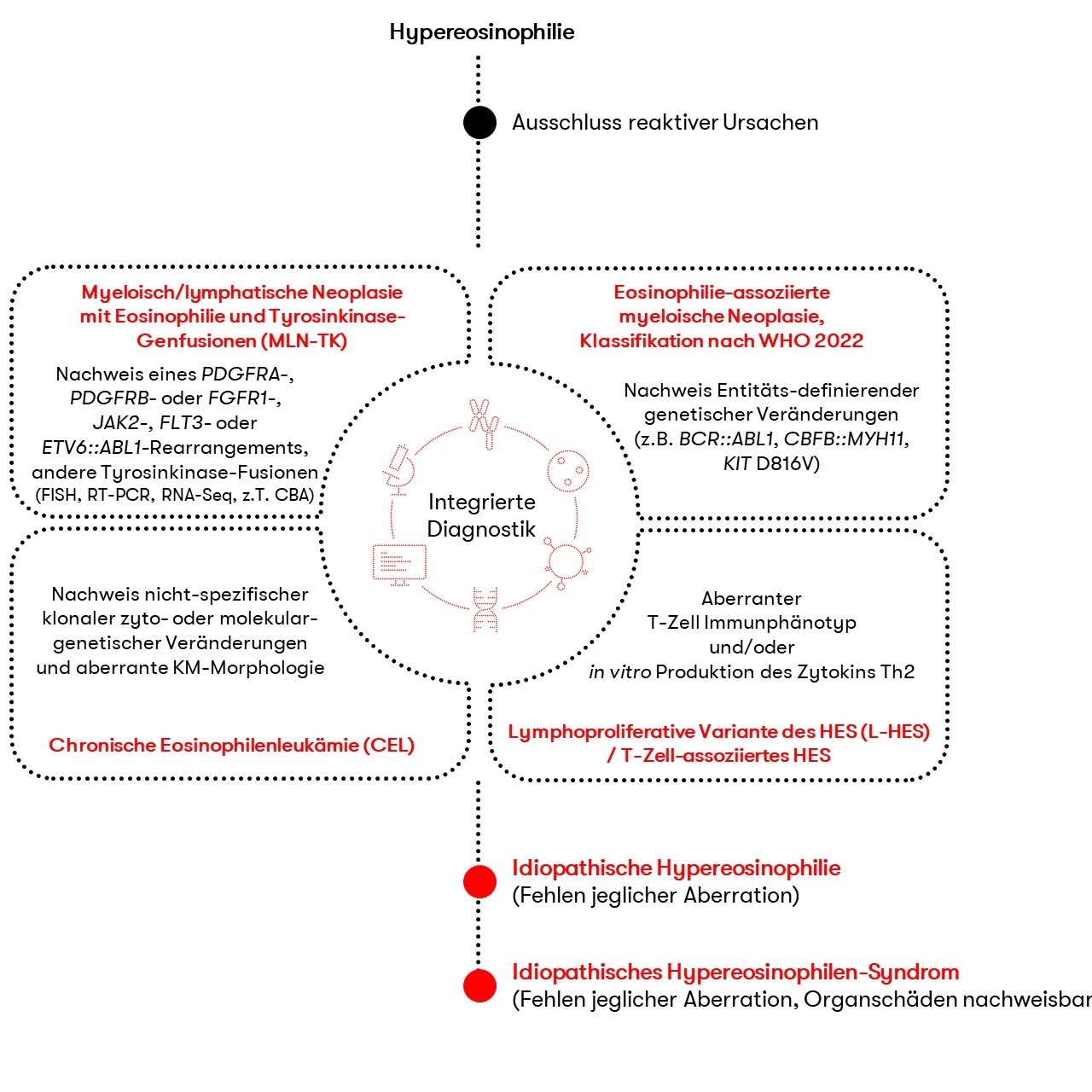
CEL: Diagnostische Methoden und ihre Bedeutung
CEL: Prognose und Therapie
Für die chronische Eosinophilenleukämie (CEL) ist eine ungünstige Prognose beschrieben und eine Transformation in eine akute Leukämie ist möglich. Laut WHO gelten ein abnormer Karyotyp, atypische Megakaryozyten, Thrombozytopenie und Knochenmarkfibrosen als potentiell ungünstige Prognosemarker (WHO 2022).
Da die CEL eine sehr seltene Entität darstellt und die Diagnose teilweise schwer zu stellen ist, umfassen bisherige Studien lediglich eine kleine Anzahl an Patienten, weshalb keine genauen Prognosefaktoren und standardisierte Behandlungsstrategien zur Verfügung stehen. Eine Übersicht über mögliche Ansätze gibt die Onkopedia Leitlinie Eosinophilie: Primäre klonale Eosinophilie und Differentialdiagnosen.
Stand: Dezember 2023