Reaktiv versus neoplastisch – Neues Panel zur Abklärung klonaler T-Zellpopulationen
Reife T-Zellneoplasien stellen in ihrer klinischen, histopathologischen und molekularen Heterogenität eine große diagnostische und therapeutische Herausforderung dar. Im Gegensatz zu den häufiger vorkommenden reifen B-Zellneoplasien, bei denen die Klonalität durch eine Restriktion der Immunglobulin-Leichtketten zu erkennen ist, gibt es bei den meisten reifen T-Zellneoplasien keine spezifische immunphänotypische Signatur. Derzeit wird beim Nachweis einer suspekten reifen T-Zellpopulation mittels Immunphänotypisierung zur weiteren diagnostischen Abklärung und zur Abgrenzung gegenüber reaktiven Veränderungen eine molekulargenetische Untersuchung zur Bestimmung der Klonalität von T-Zellrezeptor-Rearrangements empfohlen. Ein positiver Befund ist hier jedoch kein sicheres Malignitätskriterium, da nicht-maligne klonale Expansionen von T-Zellen als überschießende Immunreaktion, z. B. bei Infektionen, Autoimmunität oder auch nach Verabreichung bestimmter Medikamente, vorkommen können. In der Routinediagnostik sind daher zusätzliche genetische Analysen von aussagekräftigeren Markern erforderlich, die eine klare Unterscheidung zwischen benignen und malignen klonalen T-Zell-Populationen ermöglichen und somit die Zeit bis zur Diagnose verkürzen.
Next Generation Sequencing bei Patient:innen mit klonaler T-Zellpopulation
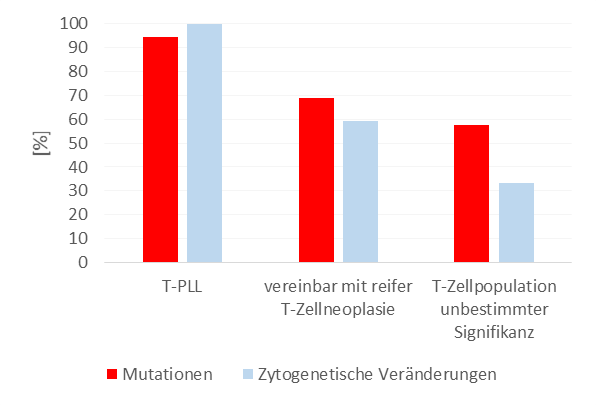
Um den potentiellen Nutzen genetischer Analysen bei Patient:innen mit suspekter reifer T-Zellpopulation zu evaluieren, wurde bei 83 Patient:innen NGS mit einem lymphatischen Gen-Panel durchgeführt. Alle Patient:innen waren zuvor mittels Immunphänotypisierung untersucht worden und für 52 Patient:innen waren zytogenetische Daten verfügbar. Basierend auf den Ergebnissen der Immunphänotypisierung wiesen 18 Patient:innen eine T-PLL auf, bei 32 Patient:innen war der Befund vereinbar mit einer sonstigen reifen T-Zellneoplasie (TCL) und 33 Patient:innen zeigten eine T-Zellpopulation unbestimmter Signifikanz (TPUS). Bei allen Patient:innen konnte in der molekulargenetischen Analyse ein klonales T-Zellrezeptor-Rearrangement detektiert werden. Die T-PLL ist aufgrund ihres spezifischen Immunphänotyps und ihrer entitätsdefinierenden zytogenetischen Veränderungen (TRAD::TCL1A-Rearrangement oder TRAD::MTCP1-Rearrangement) vergleichsweise leicht zu diagnostizieren – alle 18 Patient:innen zeigten die T-PLL-typischen zytogenetischen Veränderungen und 94% der Fälle wiesen zusätzlich Mutationen auf. Strukturelle Veränderungen und/oder Kopienzahlveränderungen wurden mittels Chromosomenanalyse und FISH zudem bei 59% der TCL- und 33% der TPUS-Patient:innen nachgewiesen. Mutationen zeigten sogar 69% der TCL- und 58% der TPUS-Patient:innen. Während Mutationen in JAK3, ATM, JAK1 und STAT5B hauptsächlich bei T-PLL auftraten, wurden Mutationen in STAT3, TET2 und DNMT3A vor allem bei TCL und TPUS beobachtet. STAT3 Mutationen treten allgemein bei ca. 30-40% der T-LGLs auf, kommen jedoch auch bei anderen reifen T-Zellneoplasien vor. Beachtlich ist, dass 60% der TPUS-Patient:innen zytogenetische und/oder molekulargenetische Marker aufwiesen.
Inzwischen sind zahlreiche Publikationen erschienen, die die molekulare Landschaft der verschiedenen T-Zellneoplasien detailliert beschreiben. Mit größeren Datensätzen und erweiterten Sequenzierungsansätzen wird der diagnostische und prognostische Wert bestimmter molekularer Veränderungen damit immer relevanter. Am bemerkenswertesten ist die hohe Anzahl sich überlappender Mutationen in epigenetischen Modifikatoren beim angioimmunoblastischen T-Zelllymphom (AITL), welches gemäß der WHO Klassifikation von 2022 als nodales follikuläres T-Helferzelllymphom vom angioimmunoblastischen Typ (nTFHL-AI) bezeichnet wird (Aleggio et al. Leukemia 2022). Dazu gehören TET2 (50%-80%), DNMT3A (20%-30%) und IDH2-R172 (20%-30%). Auch die RHOA-G17V-Mutation wird beim nTFHL-AI bei bis zu 70 % der Fälle beobachtet. Keine dieser Mutationen ist jedoch spezifisch für das nTFHL-AI, da sie auch bei anderen Entitäten beobachtet werden können, insbesondere beim nodalen PTCL mit TFH-Phänotyp (nach neuer WHO Klassifikation nTFHL-NOS). Zu den zytogenetischen Veränderungen mit diagnostischem Wert gehören ALK-Rearrangements beim ALK-positiven anaplastisch großzelligen Lymphom (ALCL), Rearrangements von DUSP22 oder TP63 beim ALK-negativen ALCL sowie ITK::SYK-Fusionen beim follikulärem T-Zelllymphom (nach neuer WHO Klassifikation nTFHL-F).
Klinische Relevanz molekulargenetischer Veränderungen
Während mit Brentuximab Vedotin bisher nur für das CD30-positive anaplastisch großzellige Lymphom (ALCL) eine effektive zielgerichtete Therapie zur Verfügung steht, werden derzeit in klinischen Studien vor allem Histon-Deacetylase-Inhibitoren (HDACi) getestet. Insbesondere R/R T-Zellneoplasien mit TFH-Phänotyp zeigten ein signifikant besseres Ansprechen im Vergleich zu T-Zellneoplasien ohne TFH-Phänotyp (HR: 0,322; p = 0,009). Diese wiesen zudem häufiger ein typisches Mutationsmuster aus TET2 und/oder DNMT3A und/oder RHOA Mutationen auf (83% vs. 40%; p = 0,034) (Ghione et al. Blood Adv 2020).
Auf Basis dieser Daten und der klinischen Relevanz erscheint eine erweiterte genetische Abklärung von klonalen T-Zellpopulationen erfolgversprechend. Deshalb haben wir unser Untersuchungsspektrum erweitert und bieten in der Molekulargenetik ein neues T-Zell-spezifisches Panel inklusive der Gene TET2, DNMT3A, RHOA-G17V, IDH2-R172, CD28, FYN, PLCG1 und VAV1 an (siehe unseren aktuellen Untersuchungsauftrag bzw. unser digitales Order Entry-System).