Myelodysplastische Neoplasie (MDS)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:obligat
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
Auf Basis der aktuellen Leitlinien und des aktuellen Forschungsstandes ergeben sich verschiedene diagnostische Empfehlungen für Patienten mit myelodysplastischer Neoplasie. Wir haben Ihnen die wichtigsten Infos zur Klassifikation und den diagnostischen Methoden am MLL zusammengefasst. Zudem haben wir weiterführende Links zur Prognose und Therapie bei myelodysplastischer Neoplasie zusammengestellt, damit Sie sich tiefergehend informieren können.
Klassifikation bei MDS
Myelodysplastische Neoplasien (MDS, früher: Myelodysplastische Syndrome) sind erworbene klonale Knochenmarkerkrankungen, die bevorzugt im höheren Lebensalter auftreten. Die MDS ist geprägt von einer proliferativen und apoptotischen Pathologie der hämatopoetischen Vorläuferzellen. Zugleich liegt häufig ein inflammatorisches Mikromilieu der hämatopoetischen Stammzellnische vor, verbunden mit einer zunehmenden Genominstabilität (Stubbins et al. 2022). Folge ist oft eine Anämie und auch Neutropenie und/oder Thrombozytopenie. Dysplasiezeichen sind in mindestens einer der drei hämatopoetischen Zelllinien zu erkennen, und es finden sich gehäuft leukämische Transformationen in eine AML. Die genetische Charakterisierung spielt eine zunehmend große Rolle in der Diagnostik, was auch in der 2022 neu erschienenen WHO-Klassifikation abgebildet wird. In dieser werden nun „MDS mit definierenden genetischen Veränderungen“ und „MDS, morphologisch definiert“ unterschieden (WHO 2022):

Die neue Aufteilung der Subgruppen soll eine genauere Klassifikation und somit die Grundlage für zielgerichtete Therapien schaffen. Da die Anzahl der dysplastischen Zelllinien in der Regel dynamisch ist und nicht unbedingt einen spezifischen MDS-Typ definiert, ist die Unterscheidung zwischen Ein- und Mehrliniendysplasie nach WHO nun optional. Innerhalb der „MDS, morphologisch definiert“ stellt zudem das „MDS, hypoplastisch (MDS-h)“ eine neue definierte Subgruppe mit Zytopenie, Dysplasie und stark verringerter Knochenmarkzellularität dar. Die Subgruppe der „MDS, nicht klassifizierbar“ wird in der neuen WHO-Klassifikation nicht mehr verwendet.
MDS: Diagnostische Methoden und ihre Bedeutung
Die Diagnose einer MDS wird durch die zytomorphologische Untersuchung von Knochenmark und ggf. peripherem Blut gestellt. Ziel ist die Abgrenzung des MDS von anderen klonalen myeloischen Erkrankungen.
Im Rahmen der zytomorphologischen Diagnostik sollten mindestens 200 (nach WHO Klassifikation sogar 500) Knochenmarkzellen und 20 Megakaryozyten evaluiert werden. Dysplasiezeichen sollten in mindestens 10% der Zellen nachweisbar sein, um die jeweilige Reihe dysplastisch nennen zu können. Einen besonderen diagnostischen Stellenwert nehmen sog. Pseudo-Pelger-Neutrophile, Ringsideroblasten, Mikromegakaryozyten und die Zahl der Blasten ein. Diese morphologischen Veränderungen korrelieren z.T. auch mit dem Vorliegen klonaler genetischer Marker. Eine solche Korrelation findet sich beispielsweise bei einer MDS mit isolierter 5q-Deletion.
Die Differenzierung zwischen hypoplastischem MDS und aplastischer Anämie ist oftmals schwierig, da sich Dysplasiezeichen in der Erythropoese bei beiden Entitäten finden können. In die Differentialdiagnose sollte auch die PNH eingeschlossen werden.
Mithilfe der Immunphänotypisierung können für MDS typische, aberrante Antigenexpressionsmuster nachgewiesen werden. Bei Patienten mit vermutetem oder gesicherter MDS kann die multiparametrische Durchflusszytometrie wertvolle Hinweise für die Diagnosestellung und auch Prognose geben. Sie ist in der Lage, aberrante Antigenexpressionsmuster auf den Zellen der Granulopoese, Monozytopoese und Erythropoese sowie auf myeloischen Progenitorzellen zu erfassen, welche mit Dysplasien korrelieren (Westers et al. 2012).
Es wird die Granularität sowie die Expression myeloischer Antigene wie beispielsweise CD11b, CD13 und CD16 erfasst. Darüber hinaus werden die myeloischen Progenitorzellen quantifiziert und auf die Expression lymphatischer und reifzelliger Marker untersucht. Auf den Monozyten wird die Expression myelomonozytärer Marker wie CD4, CD13, CD14, CD33 und CD11b beurteilt. Ferner gilt es, eine aberrante Koexpression der lymphatischen Antigene CD2 und CD56 zu erfassen. Auf den Zellen der Erythropoese kann eine aberrante Expression von CD71 festgestellt werden.
Aus den Ergebnissen der Antigenexpressionsmuster auf den einzelnen Zellreihen lässt sich ein durchflusszytometrischer Score bestimmen, dem eine prognostische Bedeutung zukommt (Wells et al. 2003).
Die Zytogenetik hat in der MDS-Diagnostik einen zentralen Stellenwert. Da für die Chromosomenanalyse teilungsfähige Zellen erforderlich sind, sollte die Chromosomenanalyse aus mit Heparin antikoaguliertem Knochenmark durchgeführt werden. Kann kein Knochenmark gewonnen werden, so kann die Analyse aus peripherem Blut versucht werden. Allerdings können im peripheren Blut die bei MDS auftretenden Chromosomenaberrationen oftmals nicht oder nur teilweise erfasst werden.
Im Falle eines aberranten Karyotyps bei zytomorphologisch nicht gesicherter Diagnose einer MDS bestätigt sich zunächst lediglich die Klonalität. Somit liegt eine klonale Hämatopoese von unbestimmtem Potential (CHIP), bzw. bei persistierender Zytopenie eine klonale Zytopenie unbestimmter Signifikanz (CCUS) vor. Bei Verlust des Y-Chromosoms handelt es sich ferner häufig um eine altersassoziierte Aberration, deren prozentualer Anteil in den Metaphasechromosomen einen prädiktiven Wert zu haben scheint. Bei einem Verlust in >75% der Metaphasen konnte in einer Studie häufiger zytomorphologisch eine MDS diagnostiziert werden, bzw. bestand eine höhere Wahrscheinlichkeit im Verlauf eine MDS zu entwickeln. Zudem wurde eine Korrelation zu MDS-assoziierten Mutationen gezeigt (Ouseph et al. 2021).
Zytogenetische Befunde können jedoch auch entitätsbestimmend sein. Die Diagnose eines „MDS-5q“ kann auch bei Vorliegen einer weiteren zytogenetischen Veränderung gestellt werden, jedoch darf es sich hierbei nicht um eine Monosomie 7 oder eine 7q-Deletion handeln. „MDS-SF3B1“ zeigen zytogenetisch keine 5q-Deletion, Monosomie 7 oder einen komplexen Karyotyp. Patienten mit MDS-biTP53 weisen häufig 17p-Deletionen und außerdem in >90% der Fälle komplexe Karyotypen auf (WHO 2022).
Die Zytogenetik ermöglicht zudem eine Einschätzung der Prognose bei Patienten mit zytomorphologisch gesichertem MDS (Bernard et al. 2022).
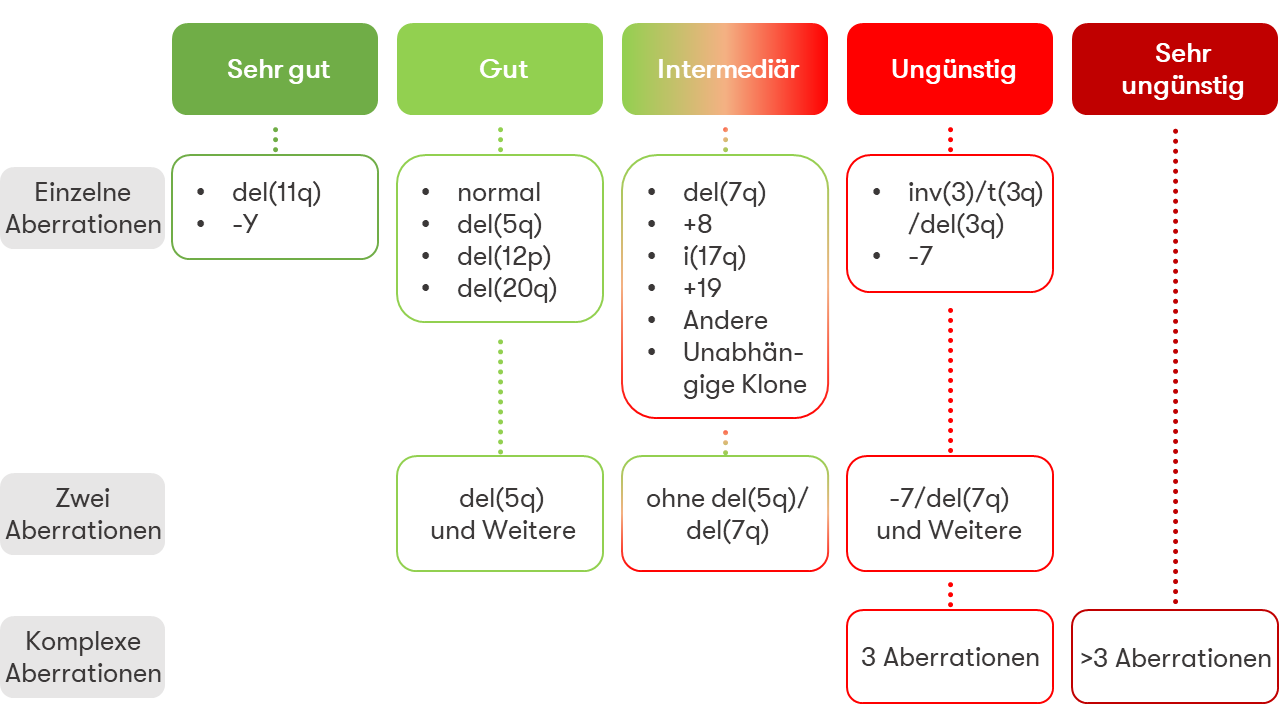
Rekurrente Chromosomenaberrationen bei MDS, z.T. mit prognostischer Relevanz nach IPSS-R bzw. IPSS-M:
- Monosomie 1, 1p-Verlust oder 1q-Zugewinn
- der(1;7)(q10;p10)
- 5q-Deletion
- Monosomie 7 oder 7q-Deletion
- Trisomie 8
- 9q-Deletion
- Trisomie 11 oder 11q-Deletion
- 12p-Deletion oder t(12p)
- Monosomie 13 oder 13q-Deletion
- 16q-Deletion
- i(17q) oder t(17p) oder 17p-Deletion
- Trisomie 19
- 20q-Deletion
- Trisomie 21
- idic(X)(q13)
- Y-Verlust
- Komplexer Karyotyp (≥3 Aberrationen)
Die prognostische Relevanz typischer Chromosomenveränderungen wird im IPSS-R in fünf zytogenetische Risikogruppen eingeteilt:
FISH dient dem Nachweis definierter genetischer Aberrationen mittels fluoreszenzmarkierter Sonden. Diese Methode kann bei der MDS zum Screening auf die häufigsten genetischen Veränderungen eingesetzt werden, wenn eine Chromosomenanalyse nicht erfolgreich durchgeführt werden konnte. Ferner können Ergebnisse aus der klassischen Chromosomenanalyse mittels FISH bestätigt werden. FISH ermöglicht zudem eine Quantifizierung des aberranten Klons am Nativmaterial und dient somit als Verlaufsmarker. Da bei einem Teil der MDS-Patienten der aberrante Klon ausschließlich im Knochenmark nachweisbar ist, stellt Knochenmark für FISH das ideale Untersuchungsmaterial dar. FISH an Zellen des peripheren Blutes ist grundsätzlich möglich.
In den letzten Jahren wurden zahlreiche Genmutationen bei MDS entdeckt, die für die Subklassifizierung, die Risikobewertung und die Charakterisierung der MDS an Bedeutung gewinnen (Papaemmanuil et al. 2013, Haferlach et al. 2014, Bernard et al. 2022). Zudem zeigte eine Studie, dass bei der Untersuchung aller proteinkodierenden Gene unter Einbeziehung von Kopienzahlvariationen alle Patienten mindestens eine genetische Veränderung aufweisen (Makishima et al. 2017), was einen enormen Fortschritt bei der genetischen Charakterisierung von MDS-Patienten widerspiegelt. Auch bei der Klassifizierung spielt die Molekulargenetik inzwischen eine bedeutende Rolle (WHO 2022).
Tabelle 1: Häufig mutierte Gene bei MDS (Bersanelli et al. 2021, Nazha et al. 2021, Bernard et al. 2022)
|
Gen |
~Häufigkeit (%) |
Prognose |
|
ASXL1* |
17-25 |
ungünstig |
|
BCOR* |
4-5 |
ungünstig |
|
BCORL1 |
bis zu 2 |
neutral |
|
CBL* |
3-5 |
ungünstig |
|
CSF3R |
<1 |
neutral |
|
DNMT3A* |
12-16 |
ungünstig |
|
ETV6 |
2-4 |
ungünstig |
|
EZH2 |
5-6 |
ungünstig |
|
FLT3 |
1 |
ungünstig |
|
GATA2 |
1 |
nicht eindeutig |
|
GNB1 |
1 |
neutral |
|
IDH1 |
3 |
ungünstig |
|
IDH2 |
4-5 |
ungünstig |
|
JAK2 |
3-4 |
unklar |
|
KMT2A PTD |
2,5 |
ungünstig |
|
KRAS |
1-3 |
ungünstig |
|
NF1 |
bis zu 3 |
ungünstig |
|
NRAS |
3-4 |
ungünstig |
|
PHF6 |
1-3 |
ungünstig |
|
PPMD1 |
bis zu 2 |
ungünstig |
|
PRPF8 |
1 |
neutral |
|
PTPN11 |
bis zu 2 |
ungünstig |
|
RAD21 |
bis zu 4 |
ungünstig |
|
RUNX1 |
8-13 |
ungünstig |
|
SETBP1 |
3 |
ungünstig |
|
SF3B1* |
20-26 |
günstig** |
|
SMC3 |
<2 |
nicht relevant |
|
SRSF2* |
12-16 |
ungünstig |
|
STAG2 |
3-9 |
ungünstig |
|
TET2* |
bis zu 31 |
günstig |
|
TP53* |
7-16 |
ungünstig*** |
|
U2AF1* |
7-9 |
widersprüchliche Daten |
|
WT1 |
bis zu 1,5 |
neutral |
|
ZRSR2 |
6 |
unklar |
*Mutationen in diesen Genen finden sich vereinzelt auch in hämatopoetischen Stammzellen gesunder Menschen (siehe „Klonale Hämatopoese von unbestimmtem Potenzial (CHIP)“) (WHO 2022).
**Die günstige Prognose gilt nur, wenn nicht gleichzeitig eine 5q-Deletion oder eine Mutation von BCOR, BCORL1, RUNX1, NRAS, STAG2 oder SRSF2 vorliegt (Bernard et al. 2022).
***In der IPSS-M Studie von Bernard konnte ein prognostisch ungünstiger Effekt nur bei >2 Veränderungen von TP53 festgestellt werden, weshalb in das IPSS-M als Risikofaktor nur TP53multihit (MDS-biTP53) eingehen.
Bei SF3B1 und TP53 handelt es sich um entitätsdefinierende Mutationen (Abb. 1). MDS-biTP53 werden durch zwei oder mehr TP53-Mutationen, eine Mutation mit nachgewiesenem TP53-Kopienzahlverlust oder Kopienzahl-neutralen Verlust der Heterozygotie definiert. Mutationen von NPM1 gelten nach der neuen WHO-Klassifikation als AML-definierende Events, daher sollten auch Fälle mit MDS-ähnlichen Merkmalen als AML mit NPM1-Mutation angesehen werden (WHO 2022). Es wurden darüber hinaus eine Reihe weiterer Genmutationen beim MDS gefunden, die allerdings sehr selten (< 5%) und außerdem überlappend mit anderen Erkrankungen auftreten und daher hier nicht weiter ausgeführt werden. Eine Übersicht über Mutationen, die in den neuen Risikoscore IPSS-M einfließen, finden Sie in der hier verlinkten Publikation (Bernard et al. 2022).
MDS: Prognose
Wesentlicher Pfeiler der Prognoseeinstufung für Patienten mit MDS war für viele Jahre das "International Prognostic Scoring System" (IPSS) (Greenberg et al. 1997). Für eine verbesserte bzw. detailliertere Risikostratifizierung der Patienten mit MDS wurde das IPSS im Jahr 2012 überarbeitet (Revised-IPSS, IPSS-R) (Greenberg et al. 2012). Die inzwischen starke Bedeutung der Molekulargenetik wird in einem neuen Prognose-Score (IPSS-M) deutlich, in dem zusätzlich zu klinischen und zytogenetischen vor allem molekulargenetische Befunde berücksichtigt werden. Die Prognoseabschätzung basiert in diesem neuen System auf dem Hämoglobinwert, der Thrombozytenzahl, der Blastenzahl im Knochenmark, der zytogenetischen Prognose-Kategorie (wie im IPSS-R) und molekulargenetischen Informationen zu 31 Genen. Eine Zusammenfassung der Informationen finden Sie in einem Übersichtsartikel auf unserer Website (Bernard et al. 2022). Hier gelangen Sie zur Prognoseberechnung des IPSS-Scores, des IPSS-R-Scores, WPSS-Scores und des neuen IPSS-M unter Berücksichtigung der molekulargenetischen Ergebnisse.
MDS: Empfehlung & Therapie
Die zytomorphologische Knochenmarkdiagnostik in Kombination mit der Zytogenetik und der Molekulargenetik stellen den aktuellen Goldstandard in der MDS-Diagnostik dar (Bernard et al. 2022). Aktuelle Studien können über das MDS-Patientenportal und die Deutsche MDS-Studiengruppe eingesehen werden.
Da MDS dynamische Erkrankungen sind, spricht sich die Onkopedia-Leitlinie für ein jährliches Screening in den Bereichen Zytomorphologie, Zytogenetik, FISH und der Molekulargenetik aus - insbesondere, wenn im individuellen Fall zu erwarten ist, dass sich eine Risikostratifikation verändern kann (Onkopedia Leitlinie MDS 2023).
Die Therapie einer myelodysplastischen Neoplasie nach deutscher Leitlinie erfolgt u.a. in Abhängigkeit der Risikogruppe sowie des Alters und des klinischen Zustands der Patienten (Onkopedia Leitlinie MDS).
Stand: September 2023