Diffus großzelliges B-Zelllymphom (DLBCL)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:fakultativ
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
Bei dem diffus großzelligen B-Zelllymphom handelt es sich um das häufigste Lymphom bei Erwachsenen. Wir haben hier die wichtigsten Infos zu (Sub-) Klassifikation, Diagnostik und Prognose zusammengefasst. Zudem haben wir weiterführende Links zur Prognose und Therapie bei DLBCL zusammengestellt, damit Sie sich tiefergehend informieren können.
DLBCL: Klassifikation
Das diffus großzellige B-Zelllymphom, nicht anderweitig klassifiziert (DLBCL, NOS) wird nach WHO-Klassifikation 2022 den reifen B-Zellneoplasien und hier der Untergruppe der großzelligen B-Zelllymphome zugeteilt. Neben dem DLBCL, NOS umfasst die WHO-Klassifikation weitere eigenständige DLBCL-Entitäten. Definierende Kriterien für diese sind die Lokalisation sowie Assoziation zu einer chronischen Inflammation oder zu bestimmten Viren (EBV, KSHV/HHV8). Bei DLBCL, NOS können MYC-, BCL2- oder BCL6-Translokationen auftreten, welche eine wesentliche Rolle für die Differentialdiagnostik spielen. Sind gleichzeitig ein MYC- und ein BCL2-Rearrangement nachweisbar, so ist ein diffus großzelliges B-Zelllymphom/hochmalignes-B-Zelllymphom mit MYC- und BCL2-Rearrangements (DLBCL/HGBL-MYC/BCL2) zu diagnostizieren. Bei gleichzeitigem Vorliegen eines MYC- sowie eines BCL6-Rearrangements ist die Zytomorphologie für die Klassifikation als DLBCL, NOS oder HGBL, NOS entscheidend (WHO 2022).
DLBCL: Subklassifikation
Morphologisch:
Anhand der Morphologie kann das DLBCL, NOS unterteilt
werden in zentroblastische, immunoblastische, anaplastische und seltene Varianten. Mit etwa 80% der Fälle ist der zentroblastische Subtyp am häufigsten (WHO 2022).
Anhand von Genexpressionsprofilen:
COO-Subtyp: Die Subtypisierung nach dem Ursprung (cell of origin, COO) ist für die exakte Klassifikation nach WHO und aufgrund ihrer prognostischen Relevanz wichtig. Unterschieden werden der GCB-Subtyp (germinal center B cells, GCB) und der ABC-Subtyp (activated B-cell, ABC) (Alizadeh et al. 2000). Circa 10-15% der Fälle sind keinem COO-Subtyp zuordenbar (Onkopedia-Leitlinie 2024).
Double-expresser Phänotyp: Der sogenannte double-expresser Phänotyp ist definiert durch die gleichzeitige Expression des MYC- und des BCL2-Gens und gilt als etablierter prognostischer Risikofaktor (Pasqualucci & Dalla-Favera 2018, WHO 2022). Circa 2/3 der Patienten mit double-expresser Phänotyp weisen ein ABC-DLBCL auf (Hu et al. 2013).
Double-hit Gensignatur: Mit dem GCB-Subtyp assoziiert ist ein Genexpressions-profil, das dem des double-hit Lymphoms (gleichzeitiges Vorliegen eines MYC- und eines BCL2-Rearrangements) ähnelt und daher als „Double-Hit Signatur“ (DHITsig) bezeichnet wird. Von double-hit Lymphomen kann das DHITsig-DLBCL durch das Fehlen gemeinsam vorliegender MYC- und BCL2-Rearrangements abgegrenzt werden. Das DHITsig-positive GCB-DLBCL zeigt gegenüber GCB-Fällen ohne diese Genexpressionssignatur eine schlechtere Prognose (Ennishi et al. 2019).
Molecular high grade: Ein weiteres mit dem GCB-Subtyp assoziiertes Genexpressionsprofil weist Merkmale des DLBCL und des Burkitt Lymphoms auf und wird als „molecular high-grade“ eingestuft (Sha et al. 2019).
Molekulare Subklassifikation
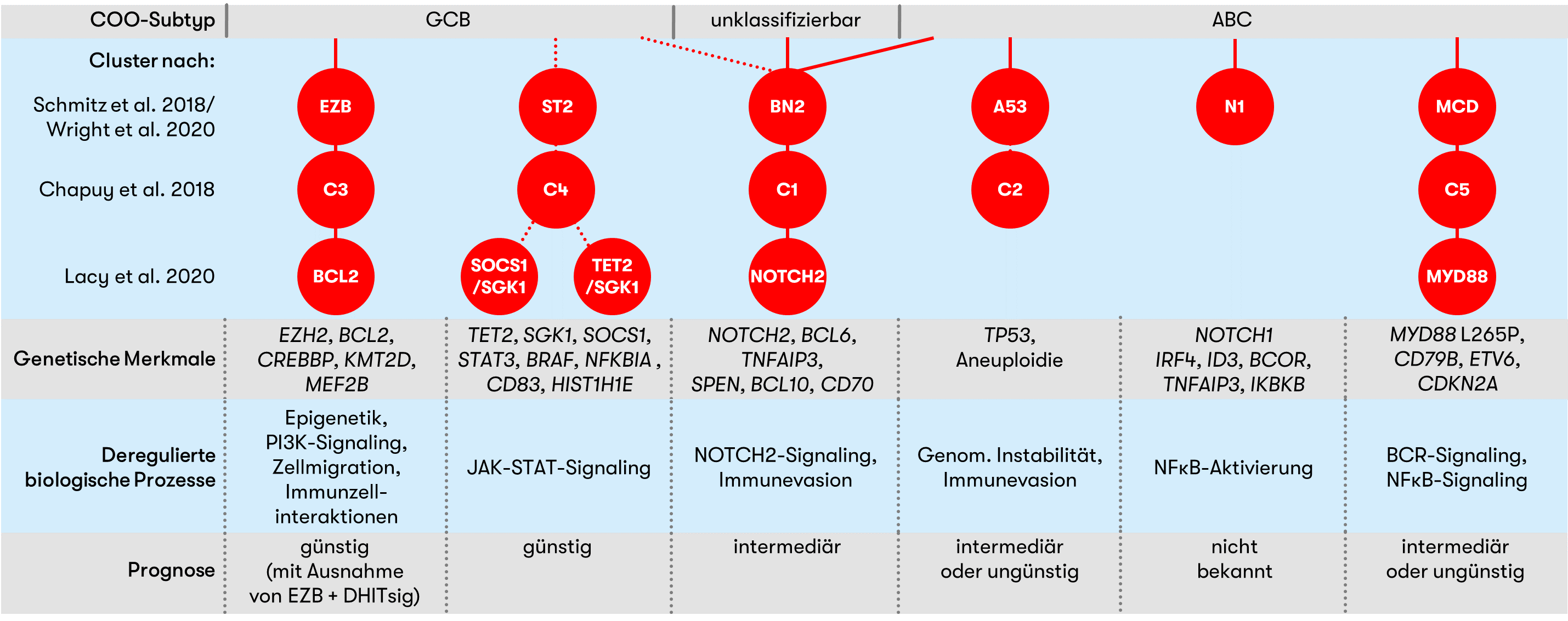
Das DLBCL ist eine genetisch heterogene Erkrankung. Dennoch zeigten verschiedene unabhängige Studien, dass sich das DLBCL anhand molekularer Signaturen in verschiedene Cluster einteilen lässt (Chapuy et al. 2018, Schmitz et al. 2018/Wright et al. 2020, Lacy et al. 2020). Diese Einteilung erlaubte in den verschiedenen Studien jeweils eine feingliedrige Risikostratifizierung und könnte zukünftig auch für die Entwicklung personalisierter Therapien von Interesse sein.
Abbildung 1 gibt einen Überblick der beschriebenen Cluster und zeigt die jeweils assoziierten molekularen Signaturen.
DLBCL: Diagnostische Methoden und ihre Bedeutung
DLBCL: Prognose
Das Genexpressionsprofil nimmt Einfluss auf die Prognose
Die Einteilung des DLBCL nach COO-Subtypen ist von prognostischer und prädiktiver Relevanz, da sich die beiden Subtypen in ihrem Ansprechen auf das R-CHOP-Regime deutlich unterscheiden. Die Prognose beim GCB-DLBCL (mit einem 5-Jahres-Überleben von ~80%) ist dabei besser als beim ABC-DLBCL (5-Jahres-Überleben von ~50%) (Pon & Marra 2016). Die gleichzeitige Expression von MYC und BCL2 (sogenannte double-expresser Phänotyp) ist mit einer ungünstigen Prognose assoziiert (Pasqualucci & Dalla-Favera 2018, WHO 2022). Hinzu treten nach aktuellen Erkenntnissen auch eine „Double-Hit“ Gen-Signatur (Ennishi et al. 2019) bzw. ein „molecular high-grade“ Genexpressionsprofil (Sha et al. 2019).
Klinische Risiko-Scores beim DLBCL
Der „Internationale Prognostische Index (IPI)“ berücksichtigt Alter, Lactatdehydrogenase-Wert, Ann-Arbor-Stadium III oder IV sowie Anzahl extranodaler Befälle. Dabei kann jeder der genannten Parameter mit einem Risikopunkt bewertet werden. Der IPI wurde vor der Rituximab-Ära eingeführt (Shipp et al. 1993). Weiterentwicklungen des IPI sind der „revised IPI“ (R-IPI), sowie der NCCN-IPI. Der R-IPI berücksichtigt auch den Performance Status (ECOG 0-1 und 3-5) (Sehn et al. 2007). Der NCCN-IPI hingegen ist bei den Faktoren Alter und LDH-Wert feingliedriger gestaltet und auch die Gewichtung unterscheidet sich im Vergleich zu dem IPI bzw. R-IPI. Bei extranodalem Befall wird zudem nicht die Anzahl, sondern die Lokalisation berücksichtigt (Zhou et al. 2014). Im Vergleich der drei klinischen Scores besaß der NCCN-IPI die größte Trennschärfe (Ruppert et al. 2020).
DLBCL: Prognoseberechnung
DLBCL: Therapie
In der Erstlinientherapie kommt das R-CHOP Regime oder R-CHOP ähnliche Protokolle zum Einsatz. Für Patienten mit erhöhtem Risiko (IPI 2-5) werden leitliniengerecht sechs Gaben R-CHP in Kombination mit Polatuzumab-Vedotin gefolgt von zwei Applikationen Rituximab empfohlen. In der Zweitlinientherapie gewinnen innovative Therapiekonzepte an Bedeutung (Onkopedia-Leitlinie 2024). Den Therapiealgorithmus nach deutscher Leitlinie finden Sie auf der Seite der Onkopedia.
Stand: April 2024