Mantelzell-Lymphom (MCL)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:fakultativ
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
Auf Basis der aktuellen Leitlinien und des aktuellen Forschungsstandes ergeben sich verschiedene diagnostische Empfehlungen für Patienten mit Mantelzell-Lymphom. Wir haben Ihnen die wichtigsten Infos zur Klassifikation und den diagnostischen Methoden am MLL zusammengefasst. Zudem haben wir weiterführende Links zur Prognose und Therapie beim Mantelzell-Lymphom zusammengestellt, damit Sie sich tiefergehend informieren können.
Mantelzell-Lymphom: Klassifikation
Beim Mantelzell-Lymphom (MCL) handelt es sich klassischer Weise um eine reife B-Zell Neoplasie mit indolenten bis aggressiven Verläufen. Charakteristisch ist die chromosomale Translokation t(11;14)(q13;q32) (IGH::CCND1) mit einer Überexpression von Cyclin D1, die in über 95% der Fälle vorliegt. Gemäß der WHO wird das Mantelzell-Lymphom (MCL) entsprechend klinisch-pathologischer Eigenschaften und zugrundeliegender pathogener Signalwege in folgende Subtypen unterteilt, wie in Abbildung 1 dargestellt (WHO 2022):
- In situ Mantelzell-Neoplasie (ISMCN)
- Mantelzell-Lymphom (MCL)
- Leukämisches nicht-nodales Mantelzell-Lymphom (nnMCL)
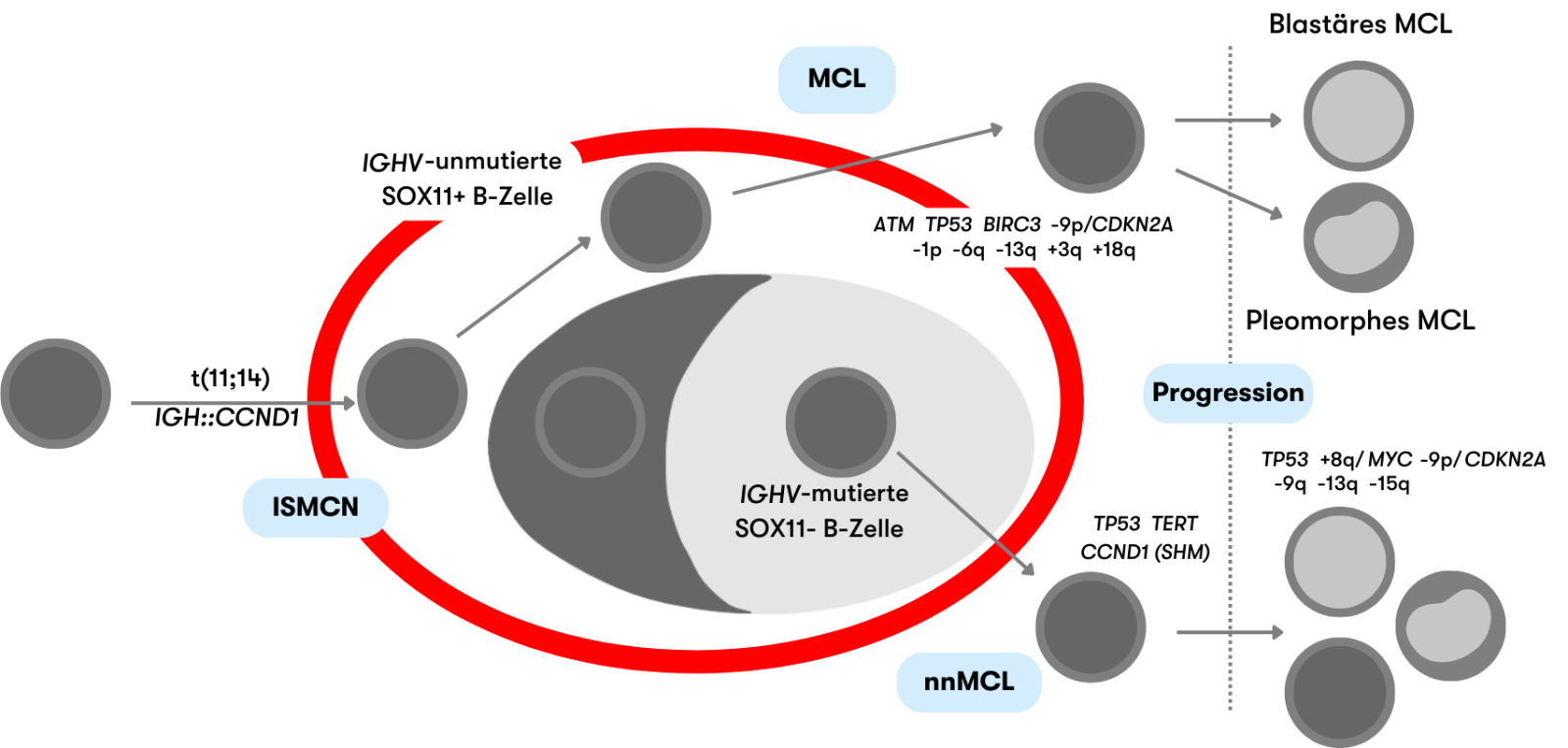
ISMCN verlaufen meist indolent und sind in der Regel Zufallsbefunde in Lymphknoten. In der Mantelzone des Lymphfollikels liegen die B-Zellen vor, die aufgrund der IGH::CCND1-Fusion eine CCND1-Überexpression aufweisen. ISMCN entwickeln sich nur selten zu MCL (<10%) (WHO 2022).
Das klassische MCL entsteht aus Zellen, die naiven reifen B-Zellen ähneln. In ≥95% der MCL liegt die IGH::CCND1-Fusion vor. MCL zeigen typischerweise eine hohe SOX11-Expression, Mutationen der Gene ATM und TP53, Kopienzahlzugewinne von 3q sowie Kopienzahlverluste von 13q und 1p. Patienten haben in der Regel eine generalisierte Lymphadenopathie und einen ungünstigen Verlauf (Nadeu et al. 2020, WHO 2022).
Bei nnMCL, die aus B-Gedächtnis-ähnlichen Zellen entstehen, liegt ebenfalls die IGH::CCND1-Fusion vor. Charakteristisch ist die Beteiligung von peripherem Blut, Knochenmark und der Milz mit geringer oder keiner Lymphadenopathie und einem meist asymptomatischen klinischen Bild. nnMCL zeigen typischerweise keine bzw. eine niedrige SOX11-Expression und weniger genetische Aberrationen als das klassische MCL (Nadeu et al. 2020, WHO 2022). SOX11-negative MCL sind die deutlich seltenere Variante und machen etwa 14%-32% der MCL aus (Cheah et al. 2016).
Mantelzell-Lymphom: Diagnostik
Mantelzell-Lymphom: Prognose
Prognostisch ungünstige Marker sind TP53-Deletionen und -Mutationen, CDKN2A-Deletionen, NOTCH1/2-Mutationen und Ki-67. TP53-Mutationen haben im Vergleich zu TP53-Deletionen einen noch schlechteren Einfluss auf die Prognose (Rubio-Moscardo et al. 2005, Salaverria et al. 2007, Sander et al. 2008, Cheah et al. 2016, Hoster et al. 2016, Eskelund et al. 2017). Das Alter, ein schlechter Allgemeinzustand, ein fortgeschrittenes Krankheitsstadium (Ann Arbor Stadium III oder IV), Splenomegalie und Anämie, der Serumspiegel von β2-Mikrogloblulin und LDH, eine blastoide Zytologie, eine extranodale Präsentation sowie konstitutionelle Symptome gehören zu wichtigen klinischen und serologischen Faktoren, die mit einem ungünstigen Verlauf assoziiert sind (Silkenstedt & Dreyling 2023).
Als klinischer Risiko-Score ist der MIPI (MCL International Prognostic Index) etabliert. In Kombination bilden der MIPI und Ki-67 Index den MIPI-c (combined MIPI) zur Risikoeinschätzung (Hoster et al. 2008, Hoster et al. 2016). Hier gelangen Sie zur Prognoseberechnung des MIPI sowie des MIPI/MIPI-c.
Mantelzell-Lymphom: Therapie
Die Wahl der Therapie sollte im Kontext mit dem Mutationsstatus getroffen werden (Martin et al. 2017). Laut der aktuellen Onkopedia Leitlinie zum Mantelzell-Lymphom, die auch detailliert über Therapiestrukturen informiert, sollten Patienten mit MCL wenn immer möglich im Rahmen von klinischen Studien behandelt werden. Dies gilt insbesondere für Patienten mit einer hohen Last an Kopienzahlveränderungen (Martin 2018).
Mantelzell-Lymphom: Empfehlung
Gemäß der aktuellen Onkopedia Leitlinie zum Mantelzell-Lymphom wird neben der Erhebung klinischer und laborchemischer Parameter aus peripherem Blut eine zytologische und histologische Untersuchung des Knochenmarks, bei leukämischem Verlauf auch eine Immunphänotypisierung der Oberflächenmarker aus peripherem Blut empfohlen.
Status: September 2023