Akute lymphatische
Leukämie (ALL)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:obligat
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
Auf Basis der aktuellen Leitlinien und des aktuellen Forschungsstandes ergeben sich verschiedene diagnostische Empfehlungen für Patienten mit akuter lymphatischer Leukämie. Wir haben Ihnen die wichtigsten Infos zur Klassifikation und den diagnostischen Methoden am MLL zusammengefasst. Zudem haben wir weiterführende Links und Literatur zur Prognose und Therapie bei ALL zusammengestellt, damit Sie sich tiefergehend informieren können.
ALL: Klassifikation
Nach der WHO-Klassifikation wird die ALL zusammen mit dem lymphoblastischen Lymphom den lymphatischen Vorläufer-Neoplasien vom B- oder T-Zell-Typ zugeordnet, wobei der Nachweis von über 25% Blasten im Knochenmark die ALL vom lymphoblastischen Lymphom abgrenzt (Swerdlow et al. 2017). Eine Einteilung in Subgruppen (Abb. 1) erfolgt primär nach zyto- und molekulargenetischen Kriterien (Alaggio et al. 2022). Klinische Relevanz hat auch die immunphänotypische Klassifikation nach EGIL, an der sich die Einteilung der immunologischen Subtypen von der GMALL-Studiengruppe orientiert, die mit spezifischen klinischen und zytogenetischen Aberrationen assoziiert sind.
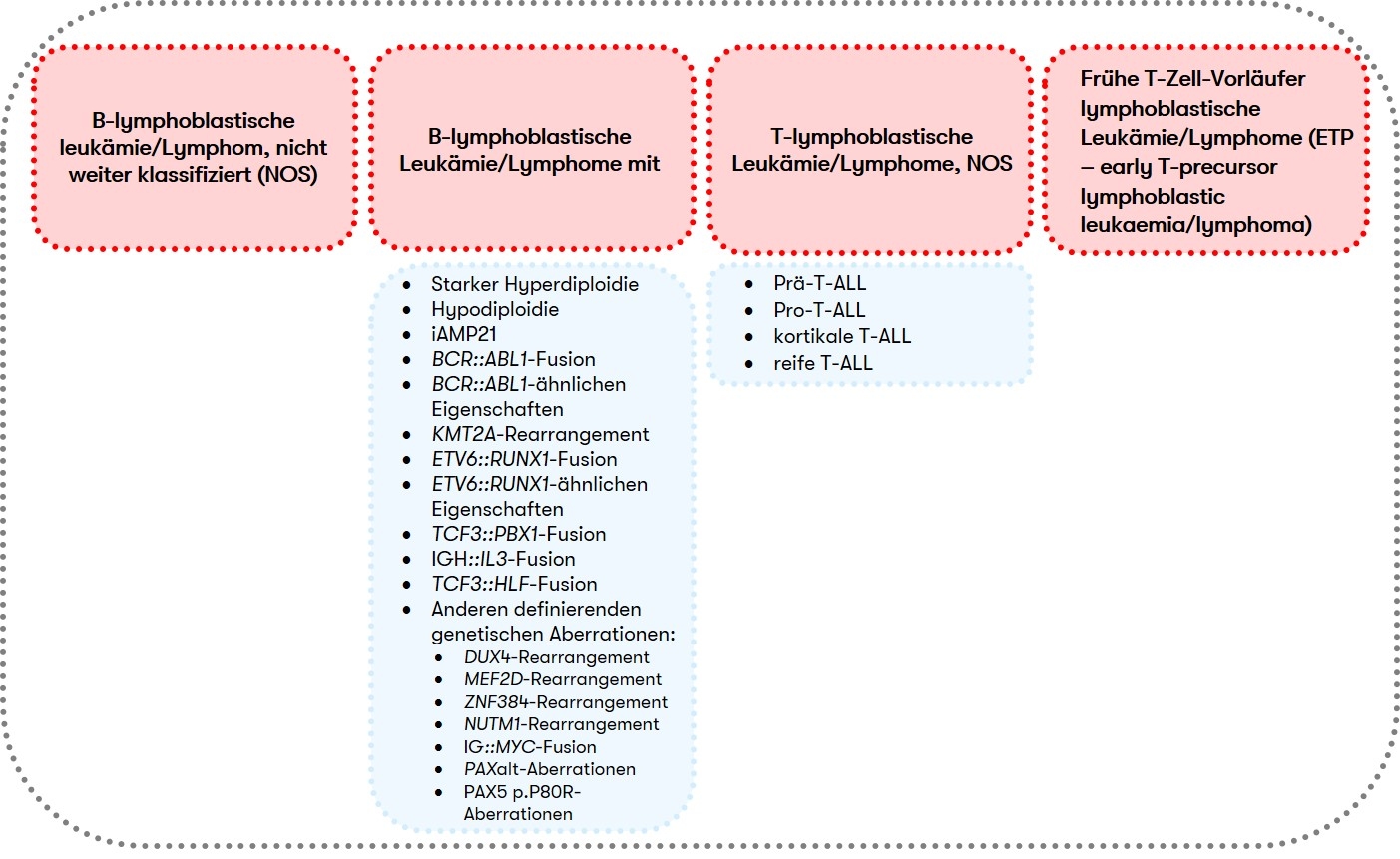
Für die B-ALL (B-lymphoblastische Leukämie/Lymphome) und T-ALL (T-lymphoblastische Leukämie/Lymphome, NOS) gelten jeweils unterschiedliche diagnostische Empfehlungen. Deshalb haben wir diese im Folgenden getrennt für Sie zusammengefasst:
B-ALL: Diagnostische Methoden und ihre Bedeutung
T-ALL: Diagnostische Methoden und ihre Bedeutung
ALL: Prognose und Therapie
Die GMALL-Studie teilt die ALL in Standard- und Hochrisikogruppen unter Berücksichtigung folgender Faktoren ein: Leukozytenzahl, Subtyp, späte CR, zyto-/molekulargenetische Aberrationen und MRD. Eine Übersicht hierzu finden Sie in der Onkopedia Leitlinie ALL.
Der MRD-Status während und nach Therapie beeinflusst das ereignisfreie Überleben und das Gesamtüberleben (Brüggemann et al. 2006, Berry et al. 2017, O’Connor et al. 2017). Die MRD ist ein hochsignifikanter Prognosefaktor und ermöglicht die frühzeitige Erkennung eines Rezidivs. Anhand ihr erfolgt die rasche Adaption der Therapiestrategie. Die Quantifizierung der MRD kann mittels Molekulargenetik oder Immunphänotypisierung durchgeführt werden.
Aufgrund der Komplexität und Langwierigkeit sollte eine Therapie in einem hämatologischen Zentrum erfolgen.
Stand: Juli 2022