Acute lymphoblastic
leukemia (ALL)
- Method:
- Anticoagulant:
- Recommendation:
- Method:Cytomorphology
- Anticoagulant:EDTA
- Recommendation:obligatory
- Method:Immunophenotyping
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
- Method:Chromosome analysis
- Anticoagulant:Heparin
- Recommendation:obligatory
- Method:FISH
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
- Method:Molecular genetics
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
Based on the current guidelines and the current state of research, there are different diagnostic recommendations for patients with acute lymphoblastic leukemia. We have summarized the most important information on classification and diagnostic methods at MLL. In addition, we provide further links and literature on prognosis and therapy in ALL, so that you can inform yourself in more detail.
ALL: Classification
According to the WHO classification, ALL, together with lymphoblastic lymphoma, is assigned to the lymphoid precursor neoplasms of the B- or T-cell type, with the detection of more than 25% blasts in the bone marrow differentiating ALL from lymphoblastic lymphoma (Swerdlow et al. 2017). Classification into subgroups (Fig. 1) is primarily based on cyto- and molecular genetic criteria (Alaggio et al. 2022). Clinical relevant is furthermore the immunophenotypic classification according to EGIL, which is the basis for the classification of immunological subtypes by the GMALL study group, which are associated with specific clinical and cytogenetic aberrations.
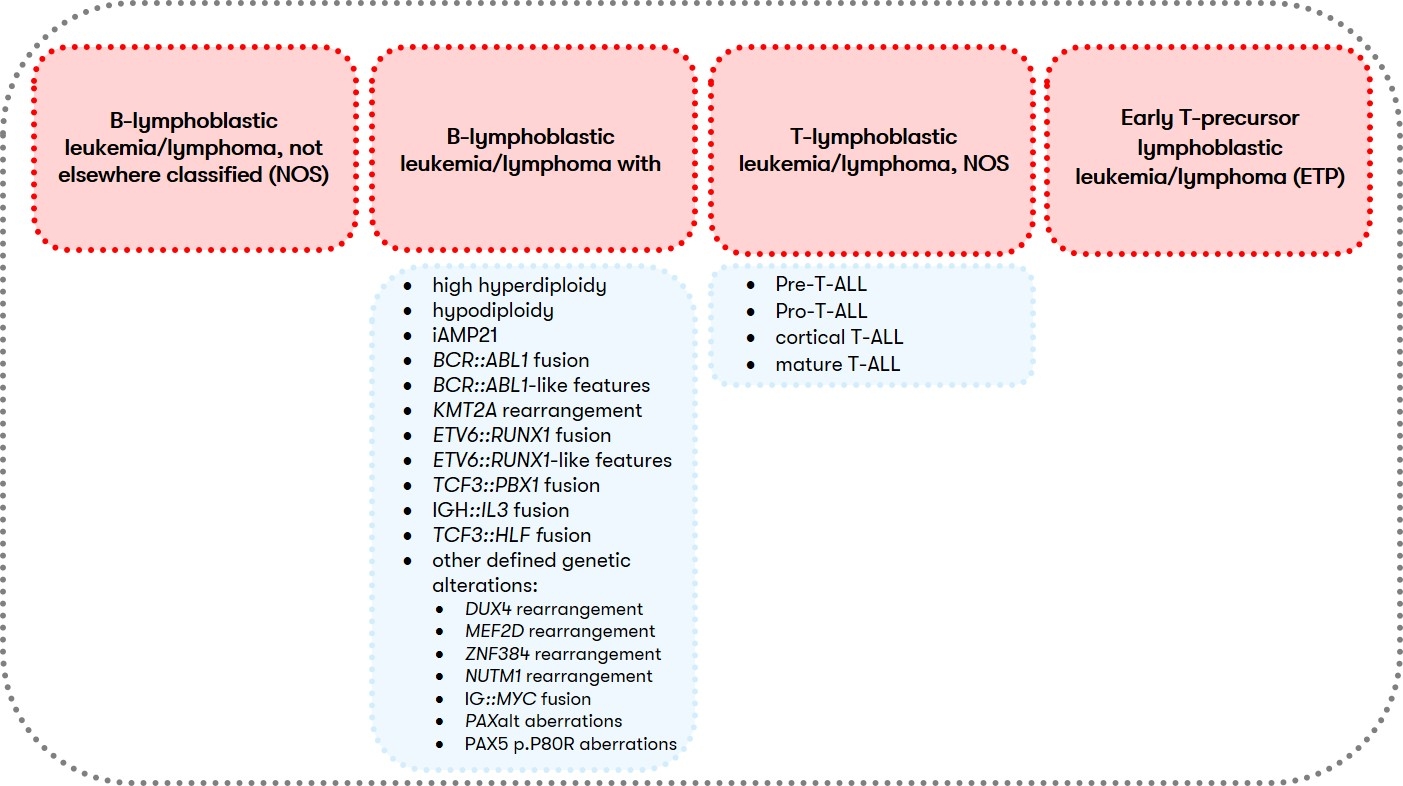
For B-ALL (B-lymphoblastic leukemia/lymphoma) and T-ALL (T-lymphoblastic leukemia/lymphoma, NOS), different diagnostic recommendations apply in each case. Therefore, we have summarized them separately for you below:
B-ALL: Diagnostic methods and their relevance
T-ALL: Diagnostic methods and their relevance
ALL: Prognosis and Therapy
The GMALL study divides ALL into standard and high-risk groups based on the following factors: Leukocyte count, subtype, late CR, cyto/molecular genetic aberrations, and MRD. For an overview, see the Onkopedia guideline ALL.
MRD status during and after therapy influences event-free survival and overall survival (Brueggemann et al. 2006, Berry et al. 2017, O'Connor et al. 2017). MRD is a highly significant prognostic factor and allows early detection of relapse. Based on it, the rapid adaptation of the therapeutic strategy takes place. Quantification of MRD can be performed by molecular genetics or immunophenotyping.
Due to the complexity and longevity, therapy should be performed in a hematological center.
Status: July 2022