MDS-assoziierte aberrante Phänotypen beim multiplen Myelom
Seit circa einem Jahrzehnt ist bekannt, dass bei einem Teil der Patienten mit multiplem Myelom (MM) MDS-assoziierte Veränderungen im Knochenmark bei Diagnose des multiplen Myeloms detektiert werden können oder im späteren Verlauf auftreten. Diese umfassen genetische Veränderungen, wie den Nachweis einer klonaler Hämatopoese bzw. MDS-assoziierter zytogenetischer Veränderungen, sowie MDS-typische aberrante Immunphänotypen1. Eine aktuelle Studie unterstreicht die klinische Relevanz solcher MDS-assoziierten (immun)phänotypischen Veränderungen (MDS-PA) und zeigt mögliche Wege für eine erweiterte Diagnostik beim MM auf.
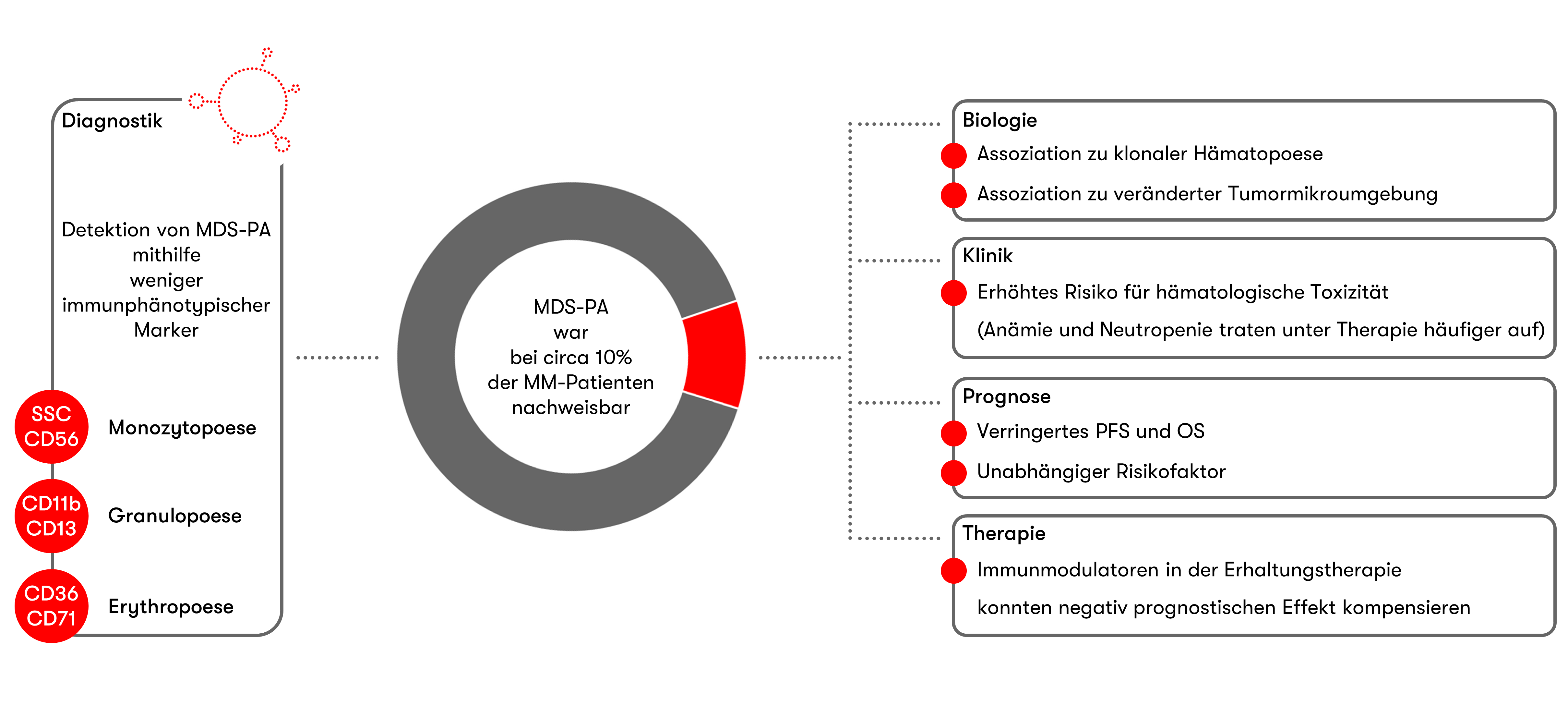
Nach ersten Literaturberichten2 zur durchflusszytometrischen Nachweisbarkeit MDS-assoziierter phänotypischer Veränderungen (MDS-PA) beim multiplen Myelom befasst sich eine aktuelle Studie (Maia et al. Blood 2020) mit der Bedeutung von MDS-PA für Biologie, Klinik, Prognose und Therapie. Daten wurden bei Diagnose sowie nach Hochdosistherapie und autologer Stammzelltransplantation (HDT/ASZT) erhoben.
Eine immunphänotypische Charakterisierung zum Zeitpunkt der Diagnose erfolgte in einer Gruppe von 285 Patienten, die an einer Therapiestudie teilnahmen. Bei 33 Fällen (11,6%) konnten im Knochenmarkaspirat MDS-typische Aberrationen nachgewiesen werden, am häufigsten war dabei eine granulozytäre Dysplasie (22 Patienten), gefolgt von erythroider (10 Patienten) und monozytärer (7 Patienten) Dysplasie. Lediglich bei 5 Fällen war mehr als eine Linie betroffen. Knochenmarkausstriche waren jedoch auch in Fällen mit MDS-PA morphologisch unauffällig. Hinsichtlich der Biologie zeigte sich eine Assoziation zu einer veränderten Tumormikroumgebung sowie zu einer klonalen Hämatopoese. Letztere wurde durch molekulargenetische Untersuchungen von 67 Patienten bei 50% der Fälle mit MDS-PA, aber nur bei 22% der Fälle ohne MDS-PA, detektiert. Die bei Diagnose detektierten phänotypischen Veränderungen bzw. die klonale Hämatopoese persistierten bei der Mehrheit der Patienten im weiteren Verlauf und nur bei einem kleinen Teil der Fälle traten MDS-PA bzw. somatische Mutationen nach HDT/ASZT neu auf.
Um die klinischen Folgen von MDS-PA evaluieren zu können, wurde eine größere Gruppe von 1.252 Patienten aus insgesamt vier Studien untersucht. Da über alle Studienkohorten hinweg nur Daten zur CD56-Expression erhoben wurden, beschränkte sich der MDS-PA-Nachweis auf die Monozyten-Linie. MDS-PA wurde bei 70 Patienten (5,6%) bei Diagnose detektiert. Patienten mit MDS-PA hatten ein erhöhtes Risiko für eine therapiebedingte hämatologische Toxizität, so traten Anämie und Neutropenie signifikant häufiger auf als bei Patienten ohne MDS-PA. Zudem bestand eine Assoziation zu einem verringerten progressionsfreien- und Gesamtüberleben. MDS-PA stellte dabei einen gegenüber etablierten Risikoparametern (ISS Stadium III, erhöhter LDH-Wert, Hochrisikozytogenetik) unabhängigen Risikofaktor dar. Dem negativ prognostischen Effekt auf das Überleben konnte jedoch durch eine post-ASZT Erhaltungstherapie mit Immunmodulatoren entgegengewirkt werden.
Zusammenfassend stellt das von Maia et al. vorgeschlagene durchflusszytometrische Screening eine kosteneffiziente und schnelle Methode dar, bereits bei Diagnose Patienten zu identifizieren, die eine dysplastische Hämatopoese aufweisen. Dies kann laut Autoren insbesondere für MM-Patienten mit ungeklärter Zytopenie sinnvoll sein. Das erhöhte Risiko für eine therapiebedingte hämatologische Toxizität und der negativ prognostische Effekt, dem therapeutisch durch die Gabe von Immunmodulatoren entgegengesteuert werden kann, macht die MDS-PA-Diagnostik klinisch relevant.
Referenzen
1Barlogie et al. Blood 2008, Usmani et al. Blood 2013, Chitre et al. Leukemia 2018
2Matarraz et al. Haematologica 2012, Matarraz et al. Leukemia 2014