Aplastic anemia (AA)
- Method:
- Anticoagulant:
- Recommendation:
- Method:Cytomorphology
- Anticoagulant:EDTA
- Recommendation:obligatory
- Method:Immunophenotyping
- Anticoagulant:EDTA or Heparin
- Recommendation:facultative
- Method:Chromosome analysis
- Anticoagulant:Heparin
- Recommendation:obligatory
- Method:FISH
- Anticoagulant:EDTA or Heparin
- Recommendation:facultative
- Method:Molecular genetics
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
Aplastic anemia describes rare and heterogeneous diseases leading to bone marrow insufficiency due to cytopenias. Based on the current guidelines and the current state of research, there are different diagnostic recommendations for patients with aplastic anemia. We have summarized the most important information on classification and diagnostic methods at MLL. In addition, we provide further links on prognosis and therapy in aplastic anemia, so that you can inform yourself in more detail.
Aplastic anemia: Classification
Acquired aplastic anemias (AA) are characterized by bi- or tricytopenias resulting from hypo- or aplasia in the bone marrow. Patients with aplastic anemia typically initially experience benign oligoclonal hematopoiesis due to a reduction in the stem cell pool resulting from immune-mediated pathogenesis, in which autoreactive T cells appear to play a major role. During the pathogenesis of aplastic anemia, the majority of bone marrow cells are replaced by adipocytes (Marsh et al. 2009, Ogawa 2016, Young 2018).
For the diagnosis of aplastic anemia, the exclusion of acquired cytopenias, as well as congenital syndromes with bone marrow insufficiency and hypoplastic myelodysplastic neoplasm (MDS) is essential (Marsh et al. 2003, Marsh et al. 2009, Killick et al. 2016). In all cases, diagnostics should include cytomorphology, histology, and cytogenetics from bone marrow.
Aplastic anemia can be classified into the following severity levels:
Table 1: Classification of aplastic anemia (two of three criteria must be met) (Onkopedia Guideline Aplastic Anemia 2022).
|
|
Moderately severe / non-severe AA |
Severe AA |
Very severe AA |
|
Neutrophil granulocytes |
< 1.2 G/L |
< 0.5 G/L |
< 0.2 G/L |
|
Thrombocytes |
< 70 G/L |
< 20 G/L |
< 20 G/L |
|
Reticulocytes |
< 60 G/L |
< 20 G/L |
< 20 G/L |
To diagnose very severe AA, < 0.2 G/L neutrophil granulocytes must be present and, in addition, hypocellular bone marrow must be present (cellularity < 25% or 25-50% with < 30% hematopoietic cells in the bone marrow) in severe and very severe AA, whereas evidence of hypocellular bone marrow is sufficient to diagnose moderately severe AA (Onkopedia Guideline Aplastic Anemia 2022).
If the classification is made according to presumed etiology, it results in the following distribution (Onkopedia Guideline Aplastic Anemia 2022):
- Idiopathic (> 80%)
- Triggered by medication (< 20%)
- Post-infectious (< 5%)
- Hereditary forms manifesting only in adulthood (5-15%)
Aplastic anemia: Diagnostics
Aplastic anemia: Prognosis
The risk of leukemic transformation is significantly increased when cytogenetic alterations are detected in chromosome banding analysis (Fig. 1).
Depending on the type of cytogenetic alteration, response to immunosuppressive therapy (IST) and prognosis may be worse (-7, complex karyotype, 5q syndrome) or better (+8, -13q) (Maciejewski & Selleri 2004, Kim et al. 2010).
In addition to cytogenetic alterations, molecular aberrations such as mutations in the genes ASXL1 and DNMT3A may also increase the risk of developing MDS or AML (see molecular genetics) (Kulasekararaj et al. 2014). In contrast to mutations in BCOR, BCORL1, and PIGA, patients with mutations in DNMT3A, ASXL1, TP53, and RUNX1 show poorer response to IST and poorer overall or progression-free survival (Ogawa 2016).
Aplastic anemia: Therapy
Whether therapy or a watch-and-wait approach is indicated depends on the severity of the disease and its causes. In the case of non-severe or moderately severe aplastic anemia, a watch-and-wait approach is usually recommended initially (Onkopedia Guideline Aplastic Anemia 2022). When therapy is indicated, with the help of immunosuppressive therapies (IST), approximately 50% of patients with aplastic anemia achieve remission, with significantly higher response in patients with age ≤ 67 years (p=0.002), coexistence of a PNH clone (p=0.017), and normal karyotype (p=0.024) (Maciejewski & Selleri 2004, Kim et al. 2010). Patients with mutations of the genes PIGA, BCOR and BCORL1 also respond significantly better to IST (Yoshizato et al. 2015, Ogawa 2016). Due to the rarity of the disease, it should be discussed with an expert center before starting therapy whether participation in a clinical trial is possible. An overview of the therapy structure can be found in the Onkopedia Guideline Aplastic Anemia.
Aplastic anemia: Recommendation
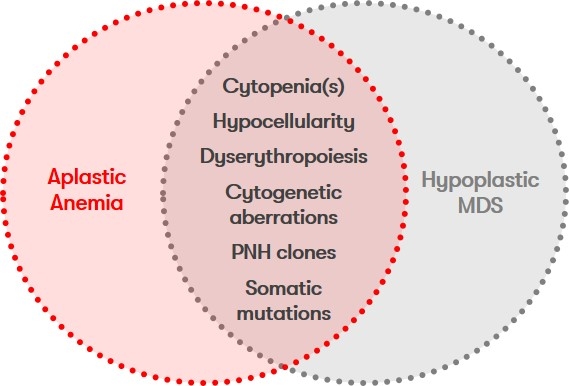
The differentiation of aplastic anemia from hypoplastic MDS is important for prognostic assessment as well as for the choice of therapeutic strategies despite the partially overlapping symptoms (Fig. 2), but morphological analyses are challenging due to the poor cellularity of the bone marrow. Therefore, histologic examination on a sufficiently large bone marrow cylinder (at least 15 mm biopsy length) should necessarily be performed to establish the diagnosis. Indications of clonal evolution in the course of aplastic anemia are an increased blast percentage, hypercellular bone marrow in the presence of recurrent or persistent cytopenia, and the appearance of new cytogenetic or molecular genetic aberrations (Maciejewski & Selleri 2004, Afable et al. 2011).
Other differential diagnoses include clonal cytopenia of undetermined significance (CCUS) and paroxysmal nocturnal hemoglobinuria (PNH).
Status: July 2023