62. ASH Annual Meeting & Exposition - a follow-up report
Also in 2020, this time as a virtual congress, the MLL was well represented at the 62nd Annual Meeting & Exposition of the American Society of Hematology (ASH) with its own presentations and in collaborations with others. Overall, the MLL and its team presented four seminars, 9 posters and one short lecture.
The focus was on the more precise characterization and classification of leukemia and lymphoma as well as the application of new methods extending to genome sequencing and artificial intelligence, both in research and in routine diagnostics.
A total of 12 scientists from the MLL presented the results from the research projects. At a cytomorphology seminar, Dr. C. Pohlkamp presented recent results from the artificial intelligence-assisted differentiation of peripheral blood counts (a project in collaboration with Amazon Web Services). Correct predictions for individual types of benign and malignant cells in peripheral blood could be made at a rate of 92% with the tested algorithm. The tools developed for this can be used in a cloud-based manner and are currently under prospective testing (BELUGA study) parallel to the gold standard of peripheral blood count differentiation by medical technicians and hematologists.
Prof. C. Haferlach then presented the current state of affairs for classic chromosomal analysis: The MLL has now succeeded in developing algorithms together with MetaSystems that can assist medical technicians and biologists with karyotyping. Well over 20,000 patients have already been processed in routine diagnostics using artificial intelligence algorithms, and this has led to a significant reduction in diagnostic times for chromosome analysis, sometimes by as much as two days compared to the previous workflow duration.
Panel sequencing with next-generation sequencing (NGS) is playing an increasingly important role in molecular genetics. For unclarified issues and excluding MDS in particular, clear benefits become evident for clinical decision-making as well as for further diagnostics and therapy selection. In her presentation, Dr. C. Bär, was able to show that with unclear constellations such as cytopenias, the analysis of a panel of genes frequently mutated in myeloid neoplasias leads to a significant improvement in the clinical diagnostic algorithms and to a reduction of differential diagnoses. For both the patient and the hematologist, this results in a reduced time to a definitive or exclusion diagnosis, while the otherwise commonly practiced multiple repeated punctures for the same clinical blood count constellation can also be reduced.
Together with ASH, the MLL has over the past two years conducted a project, whose results were presented by Mr N. Nadarajah at the ASH meeting. This involved the harmonization of molecular genetic findings, which, under the leadership of the MLL and through joint data analysis by six different laboratories in Europe and the USA, has led to a jointly agreed assessment of variants in genes that are altered in hematological diseases. The evaluation algorithms and the laboratory workflow were discussed and compared between laboratories. The result for diagnosing hematological neoplasias was published on the ASH homepage in October 2020 and is now being further developed and expanded by us.
The aim of the MLL’s scientific activities will therefore continue to improve the diagnostics for our patients, to share and make the data available for scientific publications and collaborations, and to advance new technological options such as NGS and the use of artificial intelligence to support routine laboratory diagnostics.
In the following we have summarized all the ASH projects involving MLL:
Authors: C. Baer, A. Stengel, W. Kern, C. Haferlach, T. Haferlach
Molecular genetics is playing an increasingly key role in patients with cytopenias and MDS. This is why we at the MLL have been routinely using panel sequencing for several years now. Using this data we have carried out a systematic evaluation of 576 morphologically and immunophenotypically well characterized cases along with their mutation profiles. 213 patients were classified as MDS according to the WHO criteria, of these, 85% had at least one mutation identified by panel sequencing. Mutations were also found in a third of cases without a classic MDS diagnosis. Accordingly, these are referred to as clonal cytopenia of undetermined significance (CCUS).
The aim of the study was to further subclassify CCUS patients. One quarter only showed what we refer to as DTA mutations (mutations in genes DNMT3A, TET2 or ASXL1, which often occur in old age). However, in a further 20% of the CCUS cohort, mutations in genes were found that are associated with a high risk of developing myeloid disease (e.g. RUNX1 or SF3B1). We paid particular attention to patients with SF3B1 mutations, since the international working group (IWG-PM) headed by L. Malcovati has recommended using these mutations as a criterion for diagnosing MDS, irrespective of morphology (Malcovati, et al. Blood 2020). We also found an SF3B1 mutation in 5% of CCUS patients.
Similar to these SF3B1 mutations, other genes might also play a role in the future not just in assessing the prognosis, but also in establishing the diagnosis of MDS and thereby further anchoring molecular genetics as a solid pillar of MDS diagnostics.
All information and the abstract for the study can be found here.
Authors: C. Baer*, S. Kimura*, I. Iacobucci, D. J. Feith, W. Walter, M. Meggendorfer, T. L. Olson, A. Ratan, M.-L. Mueller, M. Rana, C. Qu, R. Kalathur, C. Haferlach, W. Kern, T. P. Loughran, Jr., C. G Mullighan, T. Haferlach (*Contributed equally)
Chronic lymphoproliferative disorder of natural killer cells (CLPD-NK) is a rare hematological disorder in which a distinction between a neoplastic and a reactive state represents the greatest diagnostic challenge.
Within our whole genome sequencing program, we were able to discover previously unknown mutations in the chemokine CCL22, which occur in around a third of all patients. Together with the teams of Dr. Mullighan (St. Jude Children's Research Hospital, Memphis) and Dr. Loughran (University of Virginia School of Medicine, Charlottesville) we were able to validate these mutations in an independent cohort and carry out a number of functional studies. As one example, if cells with the mutation are transplanted into the mouse they then grow faster than control mice. In addition, the mutation also exerts an influence on the chemotactic migration of the cells in vitro.
The discovery of the CLL22 mutation has allowed us to better understand the pathogenesis of CLPD-NK, and we now have a further molecular genetic marker besides STAT3 for clonality detection in NK cell abnormalities.
You can read more information and the abstract for the study here.
Authors: C. Haferlach, S. Hänselmann, W. Walter, S. Volkert, M. Zenger, W. Kern, A. Stengel, T. Lörch, T. Haferlach
Chromosomal analysis plays an important role in the classification of numerous hematological neoplasias and in assessing their prognosis.
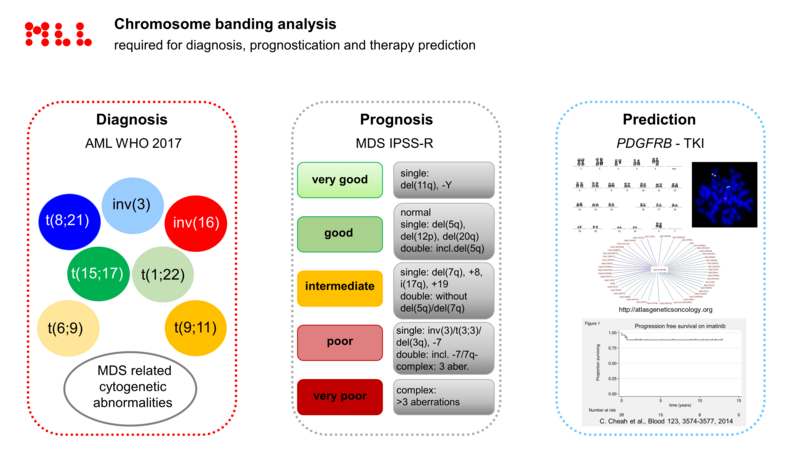
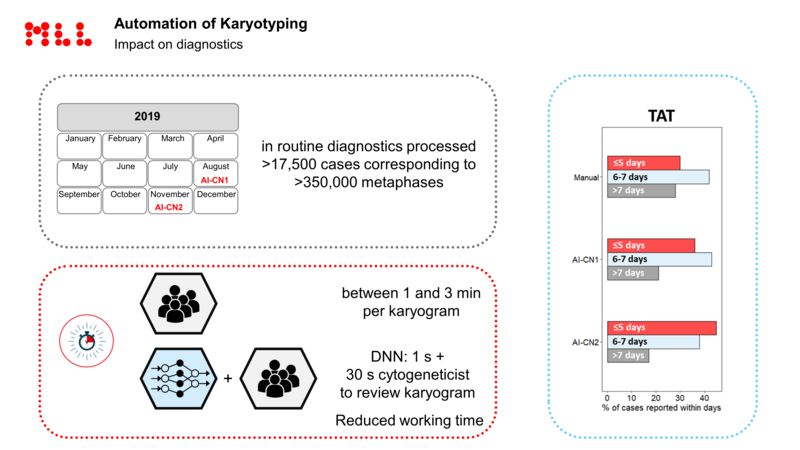
This method is very time-consuming with regard to laboratory methods and evaluation, and the quality of the results heavily depends on the experience of the cytogeneticist. Short diagnostic times are becoming more and more important as therapy decisions are increasingly being influenced by the karyotype. Laboratory processes have been automated in recent years - but now the goal has been to automate evaluation using advances in artificial intelligence (AI). A chromosome classifier was trained using 100,000 karyograms with normal karyotypes from our digital archive. A prospective validation using 500 karyograms showed a correct assignment of 22,675 of the 23,000 chromosomes (98.6%). This classifier was then introduced into clinical routine in August 2019. Since then > 350,000 metaphases have been assessed using the classifier. This has led
1) to a reduction in working time: an experienced cytogeneticist requires between 1 and 3 minutes to create a karyogram, depending on the chromosome quality, while the AI-based classifier only requires 1 second and the cytogeneticist approx. 30 seconds for validation.
Authors: K. Loy, M. Zenger, M. Meggendorfer, S. Hutter, W. Kern, T. Haferlach, C. Haferlach, C. Baer
Chronic myeloid leukemia (CML) is now a very treatable disease in the chronic phase thanks to the discovery of specific tyrosine kinase inhibitors. In contrast, patients who enter the accelerated phase or a blast crisis phase have a very poor prognosis. In order to acquire a detailed insight into the progression of individual patients with CML, 11 patients at diagnosis (D), in remission (REM) and in blast crisis (BC) were analyzed using Whole Genome Sequencing (WGS). Three different mechanisms of progression could be identified: Additional chromosomal changes besides the BCR-ABL1 translocation were detected in 91%, mutations in genes that are known to be altered in hematological neoplasias in 64%, and resistance-mediating mutations in the ABL1 of the BCR-ABL1 fusion transcript in 55% of patients. Only in the blast crisis phase could all of these changes be demonstrated. Early detection of additional changes might therefore contribute to earlier therapeutic intervention and a better prognosis.
Authors: A. Maierhofer, C. Baer, C. Pohlkamp, M. Meggendorfer, W. Kern, C. Haferlach, T. Haferlach
The screening of a cohort of 1,228 patients with sporadic AML or MDS showed that 8.3% of patients had a potential germline variant (defined as a variant with an allele frequency > 0.3) in the predisposition genes DDX41, ETV6 or GATA2. This supports the hypothesis that germline mutations in genes predisposing to myeloid neoplasias may occur more frequently than expected and emphasizes the importance of systematically testing for these variants, regardless of the patient's family history or age. 38 of the 39 patients who carried a potential germline variant in ETV6 or GATA2 had at least one other genetic variant in additionally known AML or MDS-associated genes. In 25% of the 64 patients who carried a potential germline variant in DDX41 (and in 12 cases an additional somatic DDX41 mutation), no further genetic variants could be detected in any other genes. This indicates that DDX41 and in particular the addition of a second somatic mutation promote the development of leukemia, whereas ETV6 and GATA2 seem to require additional mutations.
Authors: M. Meggendorfer, W. Walter, N. Nadarajah, C. Haferlach, W. Kern, T. Haferlach
With increasing sequencing capacities, the range of genes examined is also becoming broader. The aim of our study was therefore to determine the actual increase in genetic information with increasing NGS panel size in MDS patients, starting with small panels conforming to diagnostic guidelines up to the complete coding region (the exome). The examination of the 12 NCCN panel genes showed that 81% of MDS patients carry at least one mutation, and with extension of the analysis to the McClure panel (34 genes) this number rose to 84% and further to 87% and 100% when the Cosmic Cancer Genes (723 genes) or the exome were taken into account, respectively. In 588 patients, a total of 1,058 mutations were detected in only 12 genes. The number of mutations rose to 4,000 after examining the exome. By comparison, the number of variants of unclear significance increased from just 50 to over 21,000 when the exome was examined. This emphasizes that MDS patients can be examined with just a small diagnostic gene panel and that, examining a greater number of genes, mostly detects variants of unclear significance, whose clinical relevance needs to be determined by further studies.
All information about this project can be found on the ASH website.
Authors: L. Montefiori, S. Seliger, Z. Gz, X. Ma, B. Xu, X. Chen, K. Dickerson, T. Westover, A. Stengel, I. Iacobucci, S. Kimura, C. Qu, J. Ma, M. Valentine, W. Kern, T. Haferlach, G. Wu, J. Klco, C. Haferlach, C.G. Mullighan
Acute mixed phenotype leukemia remains difficult to diagnose and very little is known about its biological origin. In cooperation with St Jude Children's Research Hospital in Memphis, Tennessee, over 2500 samples from patients with acute leukemia were analyzed with whole transcriptome sequencing, and a subset of the patients by whole genome sequencing. A subtype of acute leukemias could be identified with T-cell and myeloid markers, which were characterized by their transcriptomic profile and their monoallelic expression of the hematopoietic transcription factor BCL11B. 80% of the cases showed changes in FLT3, and in all cases structural changes in BCL11B were detected. Most of the patients showed translocations of BCL11B with ARID1B, CCDC26, CDK6, ETV6, RUNX1 or ZEB2, which were specific for this subgroup and which were not observed in any other neoplasias. These loci contain enhancers that are active in early hematopoietic progenitor cells. In the other cases, amplification of a 2.5 kb region distal to BCL11B was detected, leading to the creation of a new enhancer – a mechanism that has never been described before. The results of this work therefore show enhancer hijacking of an otherwise line-specific transcription factor gene as a defining characteristic of a subtype of acute leukemia of unclear origin. It could also be shown that at least in some of the patients a hematopoietic stem cell is the origin of the leukemia.
Authors: J. Müller, C. Haferlach, H. Ruge, H. Müller, I. Fuhrmann, M. Meggendorfer, M.-L. Müller, W. Kern, T. Haferlach, A. Stengel)
Acute lymphoblastic T-cell leukemia (T-ALL) is a rare aggressive neoplasia that accounts for around 20% of all ALL cases. It is more common in adults than in children, although the incidence decreases with age. The subclassification of T-ALLs according to the WHO has so far been based exclusively on the immune phenotype. Although a number of frequent molecular aberrations in T-ALL have been described, a detailed molecular classification is still outstanding.
By combining Whole Genome Sequencing (WGS) and Whole Transcriptome Sequencing (WTS), we were able to subdivide 114 T-ALL cases into six different genetic subgroups at the molecular level. About half of the cases could be divided into the four already known subgroups defined by the overexpression of the oncogenes TLX1, TLX3, HOXA or TAL1 (groups 1-4). The remainder were divided into two further subgroups (groups 5 + 6) based on an oncogenic activation of the NOTCH1 signaling pathway. Here it was found that only in the last two groups (5 + 6) did mutations occur in the DNMT3A, TET2 and ASXL genes.
DNMT3A mutations in particular were detected significantly more frequently in elderly patients and are associated with a poorer overall survival. WGS analyzes also identified a previously unknown type of NOTCH1 mutation, which was designated as NOTCH1 ITD. This represents a duplication event in the NOTCH1 domain.
These data provide a basis for further classifying T-ALLs at the molecular level, identifying prognostic and MRD markers and, above all, implementing such knowledge in efficient, targeted therapies in the future.
Authors: M-L. Müller, N. Nadarajah, K. Jhalani, I. Heo, W. Wetton, C. Haferlach, T. Haferlach, W. Kern)
The use of artificial intelligence in conjunction with flow cytometric data offers major potential for the extensive automation of data evaluation. So far, this has mainly been achieved with intermediate steps using various visualization techniques, which can unfortunately be accompanied by a loss of data due to dimensional reduction. Here we were able to develop a new approach, based on the calculation of unaltered raw data. Using nine classes of B-cell lymphoma as an example, around 6400 corresponding samples were analyzed. Each of them was measured using 33 parameter flow cytometry and then evaluated both manually and with the help of artificial intelligence. In cooperation with our partner Amazon Web Services (AWS), various algorithms were developed and tested: a decision tree model, a deep learning model and an XG boost model. Considering a prediction probability of > 95% and clone sizes of the pathological entities of > 0.1%, the XG boost algorithm achieved the highest accuracy (93%) with the lowest possible proportion of unassigned cases (28%). The XG boost algorithm can be further developed in order to recognize more complex lymphoma constellations, as well as to include other pathological entities.
Authors: N. Naradajah, A. Wagner, R. Bejar, M. Ewalt, A.S. Kim, M.M. Le Beau, X. Ma, M. Meggendorfer, J.M. Shammo, G. Ryan, A.J. Siddon, D.P. Steensma, M.J. Walter, A. Zehir, J. Zhang, T. Haferlach)
Next generation sequencing (NGS) is playing an increasingly important role in diagnostics. Although the technology has made rapid advances over the past decade, the clinical interpretation of the data, and particularly the classification of somatic variants, remain problematic tasks. To help the hematology community with this problem, a working group was founded within the ASH (American Society of Hematology) led by Prof. Dr. Dr. Haferlach, which developed a reliable and generally available resource with the aim of making NGS diagnostics more consistent and robust and so improving patient care. The fruits of the work of this interdisciplinary group, consisting of hematologists, molecular biologists and bioinformaticians, is accessible as an interactive web application on the ASH homepage. The classifications of 202 variants were collated with a high level of confidence.
The original paper and the abstract for this project can be found on the ASH website.
Authors: C. Pohlkamp, K. Jhalani, N. Nadarajah, I. Heo, W. Wetton, R. Drescher, S. Hänselmann, T. Lörch, C. Haferlach, W. Kern, T. Haferlach)
Within the field of cytomorphology, the MLL presented a machine learning-based classifier for blood cells that was developed in cooperation with the companies AWS and MetaSystems. A fully automatic scanning of smears or cells from peripheral blood is first carried out. The classifier is then able to recognize and differentiate with a high level of precision all physiological and pathological cell types from digital images of peripheral blood. This also applies particularly to rare pathological cell types such as hair cells and atypical promyelocytes of APL. The tool can even assist experienced hematologists in cytomorphological diagnostics and is not restricted by the usual limitations of human examiners (concentration, fatigue, examined cell count ...). Another planned step will be the development of a classifier for bone marrow cells.
Authors: A. Stengel, M. Meggendorfer, W. Kern, T. Haferlach, C. Haferlach)
Usually, DNA damage is repaired via various DNA repair pathways. However, as a result of disruption of this equilibrium, somatic cells can accumulate mutations, which is believed to be one of the main causes not only of aging but also of cancer development. In this project, we searched for correlations between mutation frequencies (using whole genome sequencing) of 122 genes in 2656 patients with 11 different hematological neoplasias and the age of the respective patients. A total of 5709 mutations were detected in the 122 genes analyzed. A number of mutations were found that across all entities were correlated with a higher age of the patients, including so-called CHIP genes (TET2, DNMT3A, ASXL1) as well as other genes (e.g. TP53, EZH2, BCOR, GATA2, IDH2). A lower number of mutations was correlated with younger age and was partly associated with other age-related aberrations. Some mutations also showed a different age correlation depending on the entity (e.g. PHF6 mutations), suggesting different mutation or selection mechanisms depending on the respective entity.
All information and the abstract for the study can be found here.
The author

»Do you have questions regarding this article or do you need further information? Please send me an e-mail.«
Prof. Dr. med. Dr. phil. Torsten Haferlach
Executive management
Internist, Hematologist and Oncologist
Deputy Head of Cytomorphology
torsten.haferlach@mll.com