Dihydropyrimidine dehydrogenase (DPD) testing and fluorouracil toxicity
- Method:
- Anticoagulant:
- Recommendation:
- Method:Molecular genetics
- Anticoagulant:EDTA
- Recommendation:obligatory
Molecular genetic testing for DPYD variants is a diagnostic test within the meaning of § 3 No. 7 c of the German Genetic Diagnostics Act (GenDG), which requires medical education and patient consent (GEKO Guideline 2017). Therefore, the analysis can only be carried out when the declaration of consent according to GenDG signed by the patient or his legal representative is available in the laboratory.
Dihydropyrimidine dehydrogenase and fluorouracil toxicity
Fluorouracil (FU)-containing drugs are very frequently used cytostatic drugs in systemic tumor therapy. Serious and life-threatening side effects can occur in 10 - 40% of patients, and the therapy-associated lethality is 0.2 - 1.0% (Hoff et al. 2001, van Cutsem et al. 2001). A major cause of severe FU toxicity is the genetic deficiency of dihydropyrimidine dehydrogenase (DPD), the enzyme mainly responsible for FU degradation. The DPD deficiency is caused by variants in the dihydropyrimidine dehydrogenase (DPYD) gene, which are associated with an increased risk of severe, specific side effects in carriers (Meulendijks et al. 2015). In total, up to 9% of the population carries a DPYD gene variant that leads to reduced enzyme activity, and 0.1% to 0.5% show complete DPD deficiency (EMA Recommendations 2020; Amstutz et al. 2018). Therefore, pharmacogenetic testing for the most common and clinically significant DPYD gene variants is recommended prior to systemic therapy with FU-containing drugs (Lunenburg et al. 2020, DGHO Position Paper 2020).
About 30% of severe FU toxicity reactions can be explained by a genetic DPD deficiency, but there are also numerous other factors that influence the risk of severe side effects of FU-containing therapies (Amstutz et al. 2018, Froehlich et al. 2015, Schwab et al. 2008). The DGHO therefore recommends that patients with increased toxicity under FU-containing therapy not caused by the DPYD genotype should also be evaluated for other causes and, in the case of 5-FU, therapeutic drug monitoring should be performed if necessary (Wilhelm et al. 2016, DGHO Position Paper 2020).
Pharmacogenetic diagnostics of DPYD gene variants
Prediction of DPD enzyme activity based on the DPYD genotype
The prediction of the DPD phenotype on the basis of the DPYD
genotype is performed according to the DGHO guidelines, taking into
account guidelines of the Clinical Pharmacogenetics Implementation
Consortium (CPIC; Amstutz et al. 2018) and the Dutch Pharmacogenetics
Working Group (DPWG; Lunenburg et al. 2020). Essentially, a sum score of
the two weakest variant activities is formed. A score of 2 corresponds
to a normal DPD enzyme activity and a score of 0 to a complete DPD
deficiency (Table 2).
Table 2: Prediction of the DPD phenotype based on the two weakest variant activities (modified according to DGHO Position Paper 2020)
|
DPYD-Genotype |
Activity score |
|
No carrier of a DPYD variant with reduced or lost function (*1/*1) – normal enzyme activity |
2 |
|
Heterozygous carrier of a DPYD variant with reduced function (*1/c.1236G>A or *1/c.2846A>T) |
1.5 |
|
Heterozygous carrier of a DPYD variant with loss of funtion (*1/*2A or *1/*13) |
1 |
|
Carrier of two DPYD variants with reduced function (e. g. c.1236G>A and c.2846A>T) |
0.5 1,2 |
|
Carrier of a DPYD variant with reduced function and a variant with loss of funtion (combination of c.1236G>A or c.2846A>T with *2A or *13) |
0.5 1 |
|
Homozygous carrier of a DPYD variant with loss of funtion (*2A/*2A; *13/*13) or heterozygous carrier of two DPYD variants with loss of funtion (*2A/*13) |
0 |
1 Additional phenotyping is recommended for the reliable determination of enzyme activity (Lunenburg et al. 2020)
2 Different classification with an activity score of 1 according to CPIC (Amstutz et al. 2018)
There are deviations in the recommendations of the professional associations for individual constellations. For example, carriers of two DPYD variants with reduced function are classified by DGHO with a DPD activity score of 0.5, while CPIC assigns an activity score of 1 (Amstutz et al. 2018). In this constellation, the DPWG recommends additional phenotypic testing to determine DPD activity (Lunenburg et al. 2020).
Phenotypic alternatives or additions to the genetic analysis of the DPYD gene are the measurement of uracil in plasma or the physiological ratio of dihydrouracil to uracil as well as the determination of DPD activity in leucocytes (Meulendijks et al. 2016). However, the data basis for this procedure is narrower than for DPYD genetic diagnostics and the analyses are not yet part of the standard procedure in Germany before a therapy with FU-containing drugs (DGHO Position Paper 2020). In individual cases, however, such phenotypic testing may be indicated in addition to genotyping. In particular, additional phenotypic testing is recommended in the presence of two DPYD variants with reduced function or the combination of a DPYD variant with reduced function and a variant with no function (Henricks et al. 2017, Lunenburg et al. 2020).
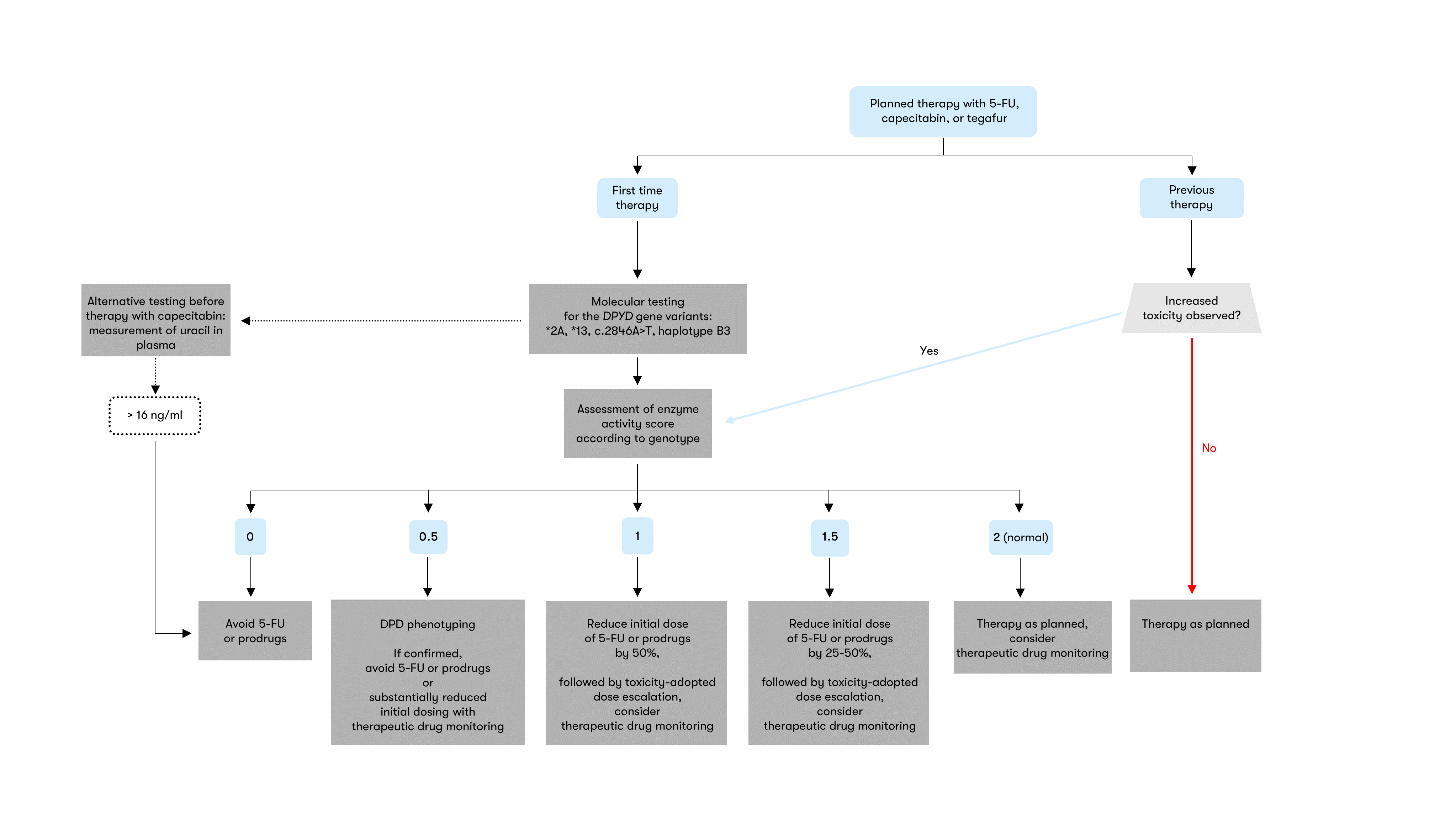
Recommendation for dosage according to DPYD genotype
The European Medicines Agency (EMA) recommends that all patients be tested for DPD deficiency prior to systemic therapy with the FU-containing drugs 5-fluorouracil (5-FU), capecitabine and tegafur (EMA Recommendations 2020). This recommendation was also taken up by the Federal Institute for Drugs and Medical Devices (BfArM) and included in the expert information of the drugs concerned. To implement these guidelines, the DGHO recommends testing for the four most common genetic DPYD variants and therapy based on a differentiated, risk-adapted algorithm according to the results of the genetic analysis, taking into account the individual disease situation and the potentially available therapy alternatives (Figure 1) (Henricks et al. 2018, DGHO Position Paper 2020). Genetic analysis can be complemented by therapeutic drug monitoring or phenotypic testing (Gamelin et al. 2008, Lunenburg et al. 2020).
Status: March 2024