Aplastische Anämie (AA)
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:obligat
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
Die Aplastische Anämie beschreibt seltene und heterogene Erkrankungen, die durch Zytopenien zu Knochenmarkinsuffizienz führt. Auf Basis der aktuellen Leitlinien und des aktuellen Forschungsstandes ergeben sich verschiedene diagnostische Empfehlungen für Patienten mit aplastischer Anämie. Wir haben Ihnen die wichtigsten Infos zur Klassifikation und den diagnostischen Methoden am MLL zusammengefasst. Zudem haben wir weiterführende Links zur Prognose und Therapie bei aplastischer Anämie zusammengestellt, damit Sie sich tiefergehend informieren können.
Aplastische Anämie: Klassifikation
Erworbene aplastische Anämien (AA) sind charakterisiert durch Bi- oder Trizytopenien, die durch Hypo- oder Aplasie im Knochenmark entstehen. Bei Patienten mit aplastischer Anämie kommt es typischerweise zunächst zu einer benignen oligoklonalen Hämatopoese durch eine Verkleinerung des Stammzellpools infolge einer immun-vermittelten Pathogenese, wobei autoreaktive T-Zellen eine große Rolle zu spielen scheinen. Während der Pathogenese der aplastischen Anämie wird der Großteil der Knochenmarkszellen durch Fettzellen ersetzt (Marsh et al. 2009, Ogawa 2016, Young 2018).
Zur Diagnose einer aplastischen Anämie ist der Ausschluss erworbener Zytopenien, sowie kongenitaler Syndrome mit Knochenmarkinsuffizienz und hypoplastischer myelodysplastischer Neoplasie (MDS) essentiell (Marsh et al. 2003, Marsh et al. 2009, Killick et al. 2016). Die Diagnostik sollte in jedem Fall Zytomorphologie, Histologie und Zytogenetik aus dem Knochenmark beinhalten.
Die aplastische Anämie kann in folgende Schweregrade eingeteilt werden:
Tabelle 1: Klassifikation der aplastischen Anämie (zwei von drei Kriterien müssen erfüllt sein) (Onkopedia Leitlinie Aplastische Anämie 2022).
|
|
Mäßig schwere / nicht-schwere AA |
Schwere AA |
Sehr schwere AA |
|
Neutrophile Granulozyten |
< 1,2 G/L |
< 0,5 G/L |
< 0,2 G/L |
|
Thrombozyten |
< 70 G/L |
< 20 G/L |
< 20 G/L |
|
Retikulozyten |
< 60 G/L |
< 20 G/L |
< 20 G/L |
Um eine sehr schwere AA zu diagnostizieren, müssen < 0,2 G/L neutrophile Granulozyten vorhanden sein und zusätzlich muss bei schwerer und sehr schwerer AA ein hypozelluläres Knochenmark vorliegen (Zellularität < 25% oder 25-50% bei < 30% hämatopoetischen Zellen im Knochenmark), während für die Diagnose einer mäßig schweren AA der Nachweis eines hypozellulären Knochenmarks reicht (Onkopedia Leitlinie Aplastische Anämie 2022).
Erfolgt die Einteilung nach vermuteter Ätiologie ergibt sich folgende Verteilung (Onkopedia Leitlinie Aplastische Anämie 2022):
- Idiopathisch (> 80%)
- Auslösung durch Medikamente (< 20%)
- Postinfektiös (< 5%)
- Sich erst im Erwachsenenalter manifestierende hereditäre Formen (5-15%)
Aplastische Anämie: Diagnostik
Aplastische Anämie: Prognose
Das Risiko einer leukämischen Transformation ist bei Nachweis zytogenetischer Veränderungen in der Chromosomenbänderungsanalyse deutlich erhöht (Abb. 1).

Das Ansprechen auf eine immunsuppressive Therapie (IST) und die Prognose können je nach Art der zytogenetischen Veränderung schlechter (-7, komplexer Karyotyp, 5q-Syndrom) oder besser (+8, -13q) sein (Maciejewski & Selleri 2004, Kim et al. 2010).
Neben zytogenetischen Veränderungen können auch molekulare Aberrationen wie Mutationen der Gene ASXL1 und DNMT3A das Risiko, ein MDS oder eine AML zu entwickeln, erhöhen (siehe Molekulargenetik) (Kulasekararaj et al. 2014). Im Gegensatz zu Mutationen in BCOR, BCORL1 und PIGA zeigen Patienten mit Mutationen in DNMT3A, ASXL1, TP53 und RUNX1 ein schlechteres Ansprechen auf eine IST sowie ein schlechteres Gesamt- bzw. Progressionsfreies Überleben (Ogawa 2016).
Aplastische Anämie: Therapie
Ob eine Therapie oder ein abwartendes Verhalten indiziert ist, hängt von der Ausprägung der Erkrankung und ihren Ursachen ab. Bei nicht schwerer oder mäßig schwerer aplastischer Anämie wird zunächst meist ein abwartendes Verhalten empfohlen (Onkopedia Leitlinie Aplastische Anämie 2022). Bei indizierter Therapie erreichen mithilfe von immunsuppressiven Therapien (IST) etwa 50% der Patienten mit aplastischer Anämie Remissionen, wobei das Ansprechen bei Patienten mit einem Alter von ≤ 67 Jahren (p=0,002), bei Koexistenz eines PNH-Klons (p=0,017) und bei normalem Karyotyp (p=0,024) signifikant höher ist (Maciejewski & Selleri 2004, Kim et al. 2010). Auch Patienten mit Mutationen der Gene PIGA, BCOR und BCORL1 sprechen signifikant besser auf IST an (Yoshizato et al. 2015, Ogawa 2016). Aufgrund der Seltenheit der Erkrankung sollte vor Therapiebeginn mit einem Expertenzentrum besprochen werden, ob die Teilnahme an einer klinischen Studie möglich ist. Eine Übersicht der Therapiestruktur ist in der Onkopedia Leitlinie Aplastische Anämie zu finden.
Empfehlung bei aplastischer Anämie
Die Abgrenzung der aplastischen Anämie zur hypoplastischen MDS ist trotz der teilweise überlappenden Symptome (Abb. 2) zur Prognoseabschätzung sowie zur Wahl der Therapiestrategien wichtig, jedoch stellen morphologische Analysen aufgrund des zellarmen Knochenmarks eine Herausforderung dar. Daher sollte zur Diagnosestellung unbedingt eine histologische Untersuchung an einem ausreichend großen Knochenmarkzylinder (mind. 15 mm Biopsielänge) durchgeführt werden. Hinweise auf eine klonale Evolution im Verlauf einer aplastischen Anämie sind ein erhöhter Blastenanteil, ein hyperzelluläres Knochenmark bei rekurrenter oder persistierender Zytopenie sowie das Auftreten neuer zytogenetischer oder molekulargenetischer Aberrationen (Maciejewski & Selleri 2004, Afable et al. 2011).
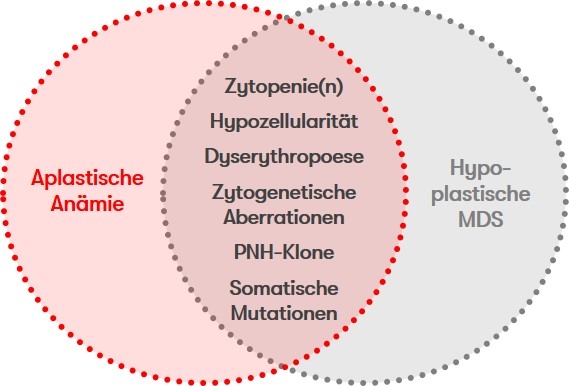
Weitere Differentialdiagnosen stellen die klonale Zytopenie unbestimmter Signifikanz (CCUS) und Paroxysmale nächtliche Hämoglobinurie (PNH) dar.
Stand: April 2024