Dihydropyrimidin-Dehydrogenase (DPD)-Testung und Fluorouracil-Toxizität
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Molekulargenetik
- Antikoagulans:EDTA
- Empfehlung:obligat
Bei der molekulargenetischen Testung auf DPYD-Varianten handelt es sich um eine diagnostische Untersuchung im Sinne von § 3 Nr. 7 c des Gendiagnostikgesetzes (GenDG), die einer ärztlichen Aufklärung und einer Einwilligung der Patienten bedarf (GEKO Richtlinie 2017). Die Analyse kann daher erst durchgeführt werden, wenn die vom Patienten bzw. dessen gesetzlichen Vertreter unterschriebene Einverständniserklärung nach GenDG im Labor vorliegt.
Dihydropyrimidin-Dehydrogenase und Fluorouracil-Toxizität
Fluorouracil (FU)-haltige Arzneimittel sind sehr häufig eingesetzte Zytostatika in der systemischen Tumortherapie. Bei 10 – 40% der Patienten können schwere und lebensbedrohliche Nebenwirkungen auftreten, die Therapie-assoziierte Letalität liegt bei 0,2 – 1,0% (Hoff et al. 2001, van Cutsem et al. 2001). Eine wesentliche Ursache für schwere FU-Toxizität ist der genetisch bedingte Mangel an Dihydropyrimidin-Dehydrogenase (DPD), dem hauptsächlich für den Abbau von FU verantwortlichen Enzym. Dem DPD-Mangel liegen Varianten im Dihydropyrimidin-Dehydrogenase-Gen (DPYD) zugrunde, welche bei den Trägern mit einem Risiko für schwere, spezifische Nebenwirkungen assoziiert sind (Meulendijks et al. 2015). In Summe tragen bis zu 9% der Bevölkerung eine DPYD-Genvariante, die zu einer verminderten Enzym-Aktivität führt, und 0,1% bis zu 0,5% weisen einen vollständigen DPD-Mangel auf (EMA Recommendations 2020; Amstutz et al. 2018). Vor einer systemischen Therapie mit FU-haltigen Arzneimitteln wird daher eine pharmakogenetische Testung auf die häufigsten und klinisch bedeutsamsten DPYD-Genvarianten empfohlen (Lunenburg et al. 2020, DGHO Positionspapier 2020).
Ca. 30% der schweren FU-Toxizitätsreaktionen sind durch einen genetischen DPD-Mangel erklärbar, es gibt jedoch auch zahlreiche weitere Faktoren, die das Risiko für schwere Nebenwirkungen FU-haltiger Therapien beeinflussen (Amstutz et al. 2018, Froehlich et al. 2015, Schwab et al. 2008). Die DGHO empfiehlt daher, bei Patienten mit erhöhter Toxizität unter FU-haltiger Therapie, die nicht durch den DPYD-Genotyp bedingt ist, auch andere Ursachen zu evaluieren und im Fall von 5-FU gegebenenfalls ein therapeutisches Drug Monitoring durchzuführen (Wilhelm et al. 2016, DGHO Positionspapier 2020).
Pharmakogenetische Diagnostik von DPYD Gen-Varianten
Vorhersage der DPD-Enzymaktivität auf Basis des DPYD-Genotyps
Die Vorhersage des DPD-Phänotyps auf Basis des DPYD-Genotyps
erfolgt nach Vorgaben der DGHO unter Berücksichtigung von Guidelines des
Clinical Pharmacogenetics Implementation Consortium (CPIC; Amstutz et
al. 2018) und der Dutch Pharmacogenetics Working Group (DPWG; Lunenburg
et al. 2020). Im Wesentlichen wird dazu ein Summenscore der beiden
schwächsten Varianten-Aktivitäten gebildet. Ein Score von 2 entspricht
dabei einer normalen DPD-Enzymaktivität und ein Score von 0 einem
kompletten DPD-Mangel (Tabelle 2).
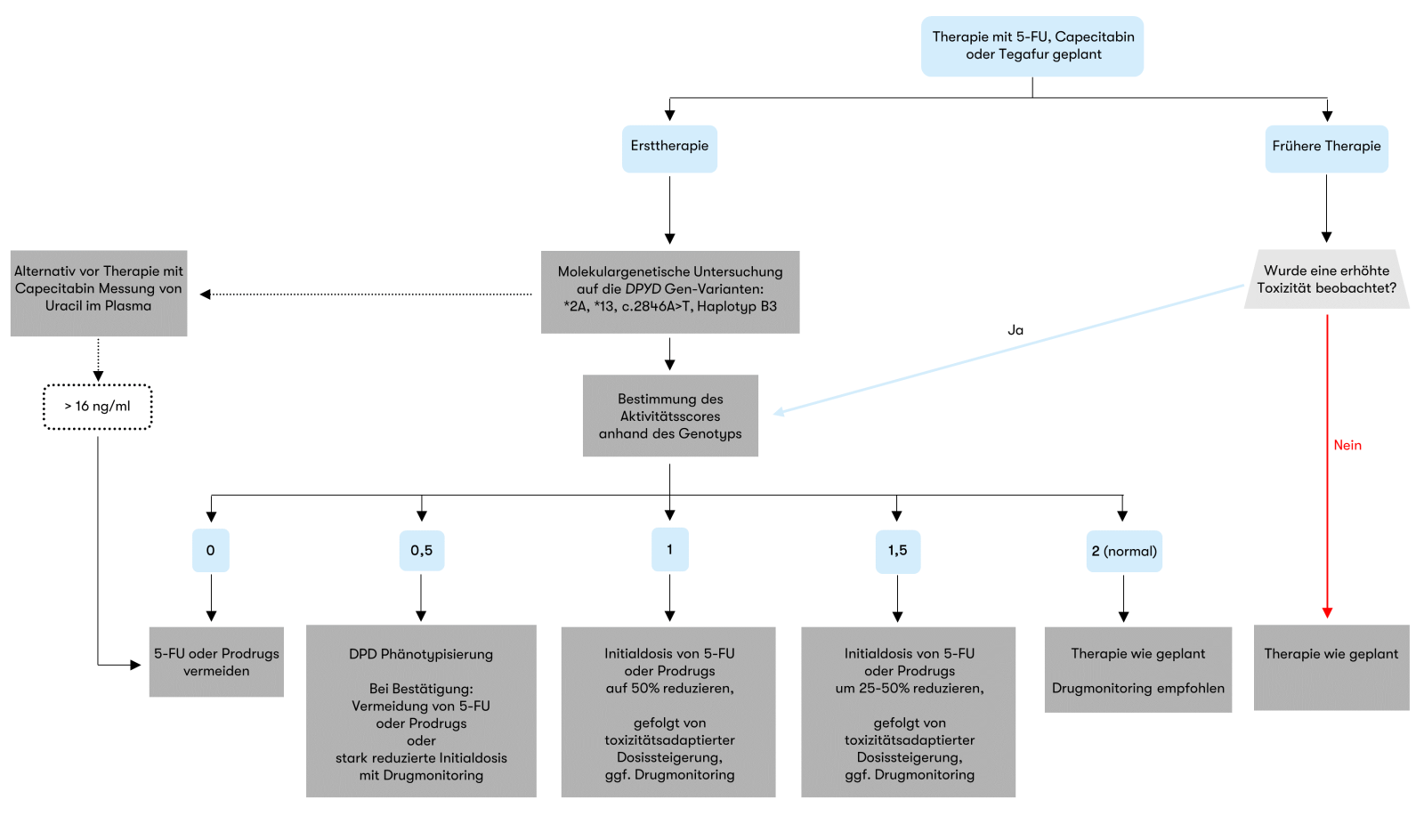
Tabelle 2: Vorhersage des DPD-Phänotyps auf der Basis der beiden schwächsten Varianten-Aktivitäten (modifiziert nach DGHO Positionspapier 2020)
|
DPYD-Genotyp |
Score der Aktivitäten
|
|
kein Träger einer DPYD-Variante mit verminderter oder fehlender Funktion (*1/*1) – normale Enzymaktivität |
2 |
|
Heterozygoter Träger einer DPYD-Variante mit verminderter Funktion (*1/c.1236G>A oder *1/c.2846A>T) |
1,5 |
|
Heterozygoter Träger einer DPYD-Variante mit fehlender Funktion (*1/*2A oder *1/*13) |
1 |
|
Träger von zwei DPYD-Varianten mit verminderter Funktion (z. B. c.1236G>A und c.2846A>T) |
0,5 1,2 |
|
Träger einer DPYD-Variante mit verminderter Funktion und einer Variante mit fehlender Funktion (Kombination von c.1236G>A oder c.2846A>T mit *2A oder *13) |
0,5 1 |
|
Homozygoter Träger einer DPYD-Variante mit fehlender Funktion (*2A/*2A; *13/*13) oder heterozygoter Träger von zwei DPYD-Varianten mit fehlender Funktion (*2A/*13) |
0 |
1 Für die sichere Festlegung der Enzymaktivität wird eine zusätzliche Phänotypisierung empfohlen (Lunenburg et al. 2020)
2 Abweichende Klassifizierung bei CPIC mit einem Aktivitätsscore 1 (Amstutz et al. 2018)
Bei einzelnen Konstellationen ergeben sich Abweichungen in den Empfehlungen der Fachgesellschaften. So werden Träger von zwei DPYD-Varianten mit verminderter Funktion nach DGHO mit einem DPD-Aktivitätsscore von 0,5 klassifiziert, während sie CPIC einem Aktivitätsscore von 1 zuordnet (Amstutz et al. 2018). Die DPWG empfiehlt bei dieser Konstellation eine zusätzliche phänotypische Testung zur Bestimmung der DPD-Aktivität (Lunenburg et al. 2020).
Phänotypische Alternativen oder Ergänzungen zur genetischen Analyse des DPYD-Gens sind die Messung von Uracil im Plasma bzw. des physiologischen Verhältnisses von Dihydrouracil zu Uracil sowie die Bestimmung der DPD-Aktivität in Leukozyten (Meulendijks et al. 2016). Die Datenbasis für dieses Vorgehen ist allerdings schmaler als für die DPYD-Gendiagnostik und die Analysen gehören in Deutschland bisher nicht zum Standardverfahren vor einer Therapie mit FU-haltigen Arzneimitteln (DGHO Positionspapier 2020). Im Einzelfall können solche phänotypischen Testungen jedoch ergänzend zur Genotypisierung indiziert sein, insbesondere wird bei Vorliegen von zwei DPYD-Varianten mit verminderter Funktion oder der Kombination einer DPYD-Variante mit verminderter Funktion und einer Variante mit fehlender Funktion eine zusätzliche phänotypische Testung empfohlen (Henricks et al. 2017, Lunenburg et al. 2020).
Empfehlung zur Dosierung nach DPYD-Genotyp
Die Europäische Arzneimittel-Agentur (EMA) empfiehlt, alle Patienten vor einer systemischen Therapie mit den FU-haltigen Arzneimitteln 5-Fluorouracil (5-FU), Capecitabin und Tegafur auf einen DPD-Mangel zu testen (EMA Recommendations 2020). Diese Empfehlung wurde auch vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) aufgegriffen und in die Fachinformationen der betroffenen Arzneimittel aufgenommen. Die DGHO empfiehlt zur Umsetzung dieser Vorgaben eine Testung auf die vier häufigsten genetischen DPYD-Varianten und eine Therapie auf Basis eines differenzierten, risiko-adaptierten Algorithmus nach Ergebnis der genetischen Analyse unter Berücksichtigung der individuellen Erkrankungssituation und der möglicherweise vorhandenen Therapiealternativen (Abbildung 1) (Henricks et al. 2018, DGHO Positionspapier 2020). Die genetische Analyse kann durch ein therapeutisches Drug Monitoring bzw. eine phänotypische Testung ergänzt werden (Gamelin et al. 2008, Lunenburg et al. 2020).
Stand: März 2024