BCR::ABL1-negative myeloproliferative Neoplasien (MPN) - Übersicht
- Methode:
- Antikoagulans:
- Empfehlung:
- Methode:Zytomorphologie
- Antikoagulans:EDTA
- Empfehlung:obligat
- Methode:Immunphänotypisierung
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Chromosomenanalyse
- Antikoagulans:Heparin
- Empfehlung:obligat
- Methode:FISH
- Antikoagulans:EDTA oder Heparin
- Empfehlung:fakultativ
- Methode:Molekulargenetik
- Antikoagulans:EDTA oder Heparin
- Empfehlung:obligat
Myeloproliferative Neoplasien (MPN) sind seltene, klonale Erkrankungen der hämatopoetischen Stammzelle, welche viele Gemeinsamkeiten aufweisen. Vor allem im Anfangsstadium lassen sie sich häufig nur schwer voneinander unterscheiden und können in einzelnen Fällen auch ineinander übergehen. Laut WHO-Klassifikation 2022 zeigt die Polyzythämia vera (PV) eine Gesamtinzidenz von 1,57/100.000 Personenjahre, die Essentielle Thrombozythämie (ET) eine Gesamtinzidenz von 1,55/100.000 Personenjahre und die Primäre Myelofibrose (PMF) im fibrotischen Stadium eine jährliche Inzidenz von 0,44-1,5 Fälle pro 100.000 Personenjahre (WHO 2022). MPN betreffen in der Regel Menschen des höheren Lebensalters mit einem Median von 60-65 Jahren (Barbui 2012). Charakteristisch bei MPN ist eine Hyperzellularität des Knochenmarks, speziell der myeloischen Zellreihe und je nach Entität eine erhöhte Anzahl von Erythrozyten, Granulozyten und/oder Thrombozyten im peripheren Blut (Haferlach et al. 2008, WHO 2022).
Myeloproliferative Neoplasien: Klassifikation
Die Einteilung in die BCR::ABL1-positive CML und die BCR::ABL1-negativen myeloproliferativen Neoplasien erfolgt gemäß der WHO-Klassifikation (Tab. 1):
Tabelle 1: MPN WHO-Klassifikation 2022 (WHO 2022)
|
Myeloproliferative Neoplasien (MPN) |
|
|
Chronische myeloische Leukämie (CML) |
BCR::ABL1-positiv |
|
Polyzythämia vera (PV) |
BCR::ABL1-negativ |
|
Essentielle Thrombozythämie (ET) |
|
|
Primäre Myelofibrose (PMF) |
|
|
Chronische Neutrophilen-Leukämie (CNL) |
|
|
Chronische Eosinophilen-Leukämie (CEL) |
|
|
Juvenile myelomonozytäre Leukämie (JMML) |
|
|
Myeloproliferative Neoplasien, nicht anderweitig klassifiziert (NOS) |
|
Myeloproliferative Neoplasien: Diagnostische Methoden und ihre Bedeutung
Myeloproliferative Neoplasien: Prognose
Neben erhöhtem Alter, Leukozytose und Thrombose (Gangat et al. 2011, Barbui et al. 2018) scheint im Allgemeinen der Nachweis chromosomaler Veränderungen bei Diagnosestellung einer MPN mit einer ungünstigeren Prognose assoziiert zu sein. Das Auftreten komplexer Karyotypen im Verlauf einer MPN erhöht die Wahrscheinlichkeit eines Übergangs in eine Blastenphase (Haferlach et al. 2008). SNP-Array Analysen zeigten in der Blastenphase gehäuft Deletionen der Gene ETV6 (Chr. 12), TP53 (Chr. 17) oder RUNX1 (Chr. 21) (Thoennissen et al. 2010).
Genmutationen beeinflussen das Risikoprofil der „klassischen“ BCR::ABL1-negativen MPN
Die JAK2 V617F-Mutation ist die häufigste Mutation bei MPN. Die PV ist fast immer mit dieser Mutation assoziiert (98%). Neben der JAK2 V617F-Mutation treten bei ET und PMF oftmals Mutationen in den Genen CALR (20-30%) und MPL (5-10%) auf (Luque Paz et al. 2023). Diese Mutationen gehören zu den Treibermutationen bei MPN, ermöglichen jedoch aufgrund ihres entitätsübergreifenden Auftretens keine spezifische Diagnose.
81% der PMF-Patienten, 53% der PV-Patienten und 53% der ET-Patienten zeigen zusätzlich zu den Treibermutationen weitere Mutationen (Nicht-Treibermutationen, Abb. 1). IDH2-Mutationen werden in allen drei klassischen BCR::ABL1-negativen MPN als Risikofaktor eingestuft (Tefferi et al. 2016, Tefferi & Vannucchi 2017). Darüber hinaus besitzt jede der drei Entitäten ein entitäts-spezifisches Risikoprofil (Abb. 1).
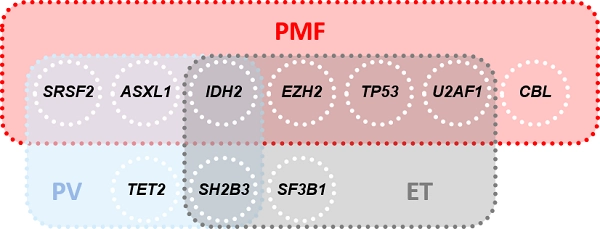
Einen detaillierten Einblick in die einzelnen MPN und deren genetische Risikoprofile finden Sie in den Infotexten der jeweiligen Entität:
Stand: Dezember 2023