Myelodysplastic neoplasms (MDS)
- Method:
- Anticoagulant:
- Recommendation:
- Method:Cytomorphology
- Anticoagulant:EDTA
- Recommendation:obligatory
- Method:Immunophenotyping
- Anticoagulant:EDTA or Heparin
- Recommendation:facultative
- Method:Chromosome analysis
- Anticoagulant:Heparin
- Recommendation:obligatory
- Method:FISH
- Anticoagulant:EDTA or Heparin
- Recommendation:facultative
- Method:Molecular genetics
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
Based on the current guidelines and the current state of research, there are different diagnostic recommendations for patients with myelodysplastic neoplasm. We have summarized the most important information on classification and diagnostic methods at MLL. In addition, we provide further links on prognosis and therapy in myelodysplastic neoplasm, so that you can inform yourself in more detail.
MDS: Classification
Myelodysplastic neoplasms (MDS, formerly: myelodysplastic syndromes) are acquired clonal bone marrow diseases that occur preferentially in older age. MDS is characterized by a proliferative and apoptotic pathology of hematopoietic progenitor cells. At the same time, an inflammatory microenvironment of the hematopoietic stem cell niche is often present, associated with increasing genome instability (Stubbins et al. 2022). Consequence is often anemia and also neutropenia and/or thrombocytopenia. Signs of dysplasia are seen in at least one of the three hematopoietic cell lineages, and leukemic transformation to AML is commonly found. Genetic characterization is playing an increasingly important role in diagnosis, which is reflected in the new WHO classification published in 2022. In this classification, "MDS with defining genetic abnormalities" and "MDS, morphologically defined" are now distinguished (WHO 2022):

The new subgrouping is intended to provide a more accurate classification and thus the basis for targeted therapies. Since the number of dysplastic cell lines is usually dynamic and does not necessarily define a specific MDS type, the distinction between single and multilineage dysplasia is now optional according to WHO. In addition, within "MDS, morphologically defined," "MDS, hypoplastic (MDS-h)" represents a newly defined subgroup with cytopenia, dysplasia, and strongly reduced bone marrow cellularity. The subgroup of "MDS, unclassifiable" is no longer used in the new WHO classification.
MDS: Diagnostic methods and their relevance
The diagnosis of MDS is made by cytomorphologic examination of bone marrow and, if required, peripheral blood. The aim is to differentiate MDS from other clonal myeloid diseases.
In the course of cytomorphological diagnostics at least 200 (according to WHO classification even 500) bone marrow cells and 20 megakaryocytes should be evaluated. Dysplasia signs should be detectable in at least 10% of the cells in order to be able to call the respective series dysplastic. So-called pseudo-Pelger neutrophils, ring sideroblasts, micromegakaryocytes and the number of blasts have a special diagnostic significance. These morphological changes correlate in part with the presence of clonal genetic markers. Such a correlation is found, for example, in MDS with isolated 5q deletion.
Differentiation between hypoplastic MDS and aplastic anemia is often difficult because signs of dysplasia in erythropoiesis can be found in both entities. PNH should also be included in the differential diagnosis.
Immunophenotyping can be used to detect aberrant antigen expression patterns typical of MDS. In patients with suspected or confirmed MDS, multiparametric flow cytometry can provide valuable information for diagnosis and also prognosis. It is able to detect aberrant antigen expression patterns on the cells of granulopoiesis, monocytopoiesis and erythropoiesis as well as on myeloid progenitor cells, which correlate with dysplasias (Westers et al. 2012).
The granularity as well as the expression of myeloid antigens such as CD11b, CD13 and CD16 will be recorded. In addition, myeloid progenitor cells are quantified and analyzed for the expression of lymphoid and mature cell markers. On the monocytes, the expression of myelomonocytic markers such as CD4, CD13, CD14, CD33 and CD11b will be assessed. Furthermore, aberrant co-expression of the lymphoid antigens CD2 and CD56 is to be detected. On the cells of erythropoiesis an aberrant expression of CD71 can be detected.
From the results of the antigen expression patterns on the individual cell series, a flow cytometric score can be determined, which has prognostic significance (Wells et al. 2003).
Cytogenetics is of central importance in MDS diagnostics. Since chromosome analysis requires cells capable of dividing, chromosome analysis should be performed from bone marrow anticoagulated with heparin. If bone marrow cannot be obtained, analysis from peripheral blood can be attempted. However, peripheral blood often fails to detect or only partially detects the chromosomal aberrations that occur in MDS.
In the case of an aberrant karyotype with cytomorphologically unconfirmed diagnosis of MDS, only clonality is initially confirmed. Thus, clonal hematopoiesis of undetermined potential (CHIP), or in case of persistent cytopenia, clonal cytopenia of undetermined significance (CCUS) is present. Furthermore, loss of the Y chromosome is often an age-associated aberration, whose percentage in metaphase chromosomes seems to have predictive value. In one study, a loss in ≥75% of metaphases resulted in a more frequent cytomorphological diagnosis of MDS or a higher probability of developing MDS during the course of the disease. In addition, a correlation to MDS-associated mutations was shown (Ouseph et al. 2021).
However, cytogenetic findings may also be entity-defining. The diagnosis of "MDS-5q" can also be made in the presence of another cytogenetic alteration, but this must not be a monosomy 7 or a 7q deletion. "MDS-SF3B1" cytogenetically do not show a 5q deletion, monosomy 7, or complex karyotype. Patients with MDS-biTP53 often have 17p deletions and also complex karyotypes in >90% of cases (WHO 2022).
Cytogenetics also allows an assessment of prognosis in patients with cytomorphologically confirmed MDS (Bernard et al. 2022).
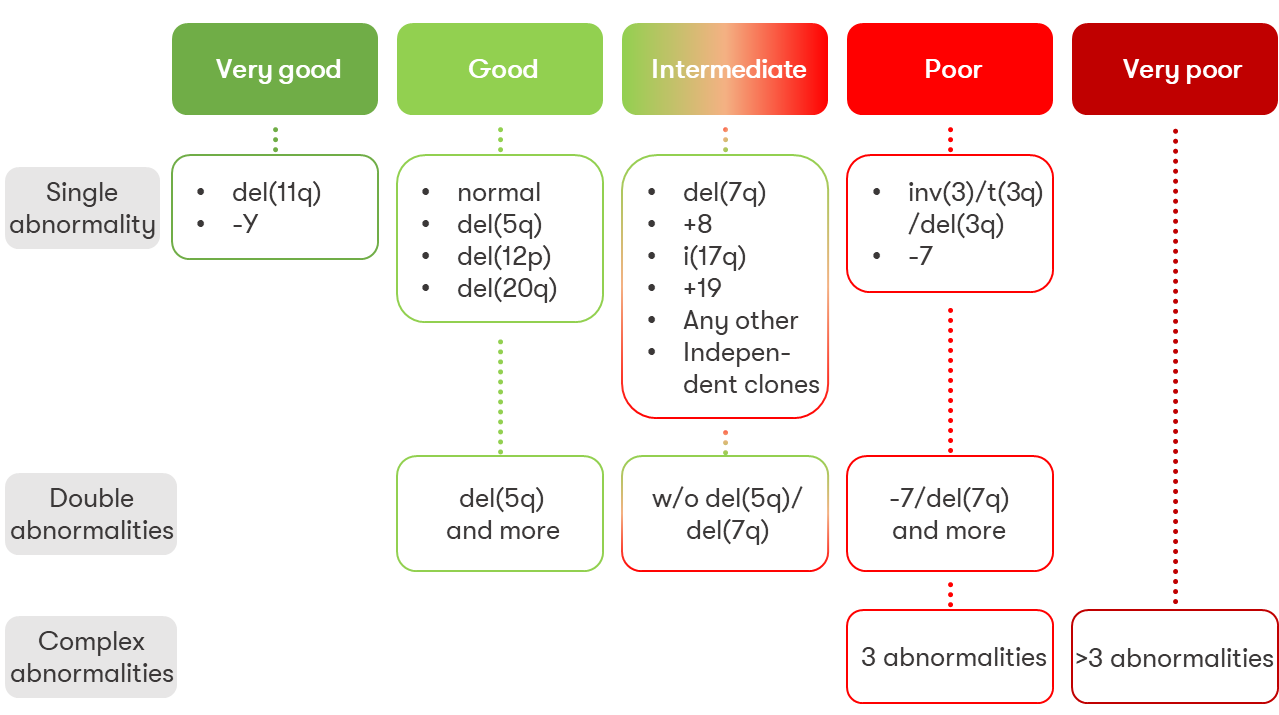
Recurrent chromosomal aberrations in MDS, partly with prognostic relevance according to IPSS-R or IPSS-M:
- monosomy 1, 1p loss or 1q gain
- der(1;7)(q10;p10)
- 5q deletion
- monosomy 7 or 7q deletion
- trisomy 8
- 9q deletion
- trisomy 11 or 11q deletion
- 12p deletion or t(12p)
- monosomy 13 or 13q deletion
- 16q deletion
- i(17q) or t(17p) or 17p deletion
- trisomy 19
- 20q deletion
- trisomy 21
- idic(X)(q13)
- Y loss
- complex karyotype (>3 aberrations)
The prognostic relevance of typical chromosomal alterations is classified into five cytogenetic risk groups in the IPSS-R:
FISH is used to detect defined genetic aberrations using fluorescently labeled probes. This method can be used in MDS to screen for the most common genetic alterations when chromosome analysis could not be successfully performed. Furthermore, results from classical chromosome analysis can be confirmed by FISH. FISH also allows quantification of the aberrant clone on native material and thus serves as a progression marker. Since in some MDS patients the aberrant clone is exclusively detectable in bone marrow, bone marrow is the ideal material for FISH. FISH on cells of peripheral blood is possible in principle.
In recent years, numerous gene mutations in MDS have been discovered, which are gaining importance for subclassification, risk assessment, and characterization of MDS (Papaemmanuil et al. 2013, Haferlach et al. 2014, Bernard et al. 2022). In addition, one study showed that when all protein-coding genes were examined including copy number variations, all patients had at least one genetic alteration (Makishima et al. 2017), reflecting tremendous progress in the genetic characterization of MDS patients. Also in classification molecular genetics plays a significant role now (WHO 2022).
Table 1: Commonly mutated genes in MDS (Bersanelli et al. 2021, Nazha et al. 2021, Bernard et al. 2022)
|
Gene |
~Frequency (%) |
Prognosis |
|
ASXL1* |
17-25 |
unfavorable |
|
BCOR* |
4-5 |
unfavorable |
|
BCORL1 |
up to 2 |
neutral |
|
CBL* |
3-5 |
unfavorable |
|
CSF3R |
<1 |
neutral |
|
DNMT3A* |
12-16 |
unfavorable |
|
ETV6 |
2-4 |
unfavorable |
|
EZH2 |
5-6 |
unfavorable |
|
FLT3 |
1 |
unfavorable |
|
GATA2 |
1 |
not clear |
|
GNB1 |
1 |
neutral |
|
IDH1 |
3 |
unfavorable |
|
IDH2 |
4-5 |
unfavorable |
|
JAK2 |
3-4 |
not clear |
|
KMT2A PTD |
2,5 |
unfavorable |
|
KRAS |
1-3 |
unfavorable |
|
NF1 |
up to 3 |
unfavorable |
|
NRAS |
3-4 |
unfavorable |
|
PHF6 |
1-3 |
unfavorable |
|
PPMD1 |
up to 2 |
unfavorable |
|
PRPF8 |
1 |
neutral |
|
PTPN11 |
up to 2 |
unfavorable |
|
RAD21 |
up to 4 |
unfavorable |
|
RUNX1 |
8-13 |
unfavorable |
|
SETBP1 |
3 |
unfavorable |
|
SF3B1* |
20-26 |
favorable** |
|
SMC3 |
<2 |
not relevant |
|
SRSF2* |
12-16 |
unfavorable |
|
STAG2 |
3-9 |
unfavorable |
|
TET2* |
up to 31 |
favorable |
|
TP53* |
7-16 |
unfavorable*** |
|
U2AF1* |
7-9 |
contradictory data |
|
WT1 |
up to 1,5 |
neutral |
|
ZRSR2 |
6 |
not clear |
*Mutations in these genes occasionally occur also in hematopoietic stem cells of healthy humans (see "Clonal hematopoiesis of undetermined potential (CHIP)") (WHO 2022).
**The favorable prognosis applies only if there is no concomitant 5q deletion or mutation of BCOR, BCORL1, RUNX1, NRAS, STAG2, or SRSF2 (Bernard et al. 2022).
***In the IPSS-M study by Bernard, a prognostically unfavorable effect could only be found with >2 alterations of TP53, which is why only TP53multihit (MDS-biTP53) is included as a risk factor in the IPSS-M.
SF3B1 and TP53 are entity-defining mutations (Fig. 1). MDS-biTP53 are defined by two or more TP53 mutations, a mutation with proven TP53 copy number loss, or copy number-neutral loss of heterozygosity. Mutations of NPM1 are considered AML-defining events according to the new WHO classification, and therefore cases with MDS-like features should also be considered AML with NPM1 mutation (WHO 2022). A number of other gene mutations have also been found in MDS, but these are very rare (<5%) and also overlap with other diseases and are therefore not discussed further here. An overview of mutations included in the new IPSS-M risk score can be found in the publication linked here (Bernard et al. 2022).
MDS: Prognosis
For many years, the International Prognostic Scoring System (IPSS) (Greenberg et al. 1997) was the mainstay of prognostic classification for patients with MDS. For improved or more detailed risk stratification of patients with MDS, the IPSS was revised in 2012 (Revised-IPSS, IPSS-R) (Greenberg et al. 2012). The now strong emphasis on molecular genetics is reflected in a new prognostic score (IPSS-M), which takes into account primarily molecular genetic findings in addition to clinical and cytogenetic ones. In this new system, prognosis assessment is based on hemoglobin level, platelet count, bone marrow blast count, cytogenetic prognostic category (as in IPSS-R), and molecular genetic information on 31 genes. A summary of the information can be found in a review article on our website (Bernard et al. 2022). Click here for the prognosis calculation of the IPSS score, the IPSS-R score, WPSS score and the new IPSS-M taking into account the molecular genetic results.
MDS: Recommendation & Therapy
Cytomorphological bone marrow diagnostics in combination with cytogenetics and molecular genetics represent the current gold standard in MDS diagnostics (Bernard et al. 2022). Current studies can be accessed via the MDS Patient Portal and the German MDS Study Group.
Since MDS are considered dynamic diseases, the Onkopedia guideline advocates annual screening in the areas of cytomorphology, cytogenetics, FISH, and molecular genetics - especially if risk stratification can be expected to change in an individual case (Onkopedia Guideline MDS 2023).
Therapy of myelodysplastic neoplasm according to the German guideline depends, among other things, on the risk group as well as the age and clinical condition of the patient (Onkopedia Guideline MDS).
Status: September 2023