Waldenström Macroglobulinemia / Lymphoplasmacytic lymphoma (LPL)
- Method:
- Anticoagulant:
- Recommendation:
- Method:Cytomorphology
- Anticoagulant:EDTA
- Recommendation:obligatory
- Method:Immunophenotyping
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
- Method:Chromosome analysis
- Anticoagulant:Heparin
- Recommendation:facultative
- Method:FISH
- Anticoagulant:EDTA or Heparin
- Recommendation:facultative
- Method:Molecular genetics
- Anticoagulant:EDTA or Heparin
- Recommendation:obligatory
Waldenström macroglobulinemia is a rare lymphoproliferative disorder characterized by the excessive production of IgM monoclonal antibodies. Based on the current guidelines and the current state of research, there are different diagnostic recommendations for patients with Waldenström macroglobulinemia. We have summarized the most important information on classification and diagnostic methods at the MLL. In addition, we provide further links on prognosis and therapy in Waldenström macroglobulinemia.
Waldenström macroglobulinemia: Classification
Waldenström macroglobulinemia is a rare chronic lymphoproliferative disorder that is usually indolent. Only 10-15% of patients show more rapid progression of the disease. According to the WHO classification of 2022, Waldenström macroglobulinemia is classified as mature B-cell neoplasms and here assigned to lymphoplasmacytic lymphomas (LPL). WHO distinguishes 2 subtypes of LPL, of which Waldenström macroglobulinemia / IgM-LPL is the far more common. Non- Waldenström macroglobulinemia LPL account for only about 5% of LPL. They include cases with IgG or IgA monoclonal proteins, non-secretory LPL, and IgM-LPL without bone marrow involvement (WHO 2022).
Characteristic and diagnosis-defining features are a lymphoplasmacytic infiltration of the bone marrow and a monoclonal immunoglobulin M (IgM). The origin of the pathological cell population are probably B cells that have not yet undergone an isotype change but have already undergone the germinal center reaction.
The laboratory diagnosis of IgM-MGUS (monoclonal gammopathy of undetermined significance) develops into Waldenström macroglobulinemia at a progression rate of 1.5-2% per year (Kyle et al. 2003, Bustoros et al. 2019). IgM-MGUS is defined by a serum monoclonal IgM concentration below 30 g/l and less than 10% plasma cells in the bone marrow (WHO 2022). 6q deletions that are characteristic of Waldenström macroglobulinemia (see also chromosomal analysis) represent a risk factor for transformation of IgM-MGUS into Waldenström macroglobulinemia (Paiva et al. 2015, Guerrera et al. 2018).
Table 1: Classification of Waldenström´s macroglobulinemia and related disorders according to the European Consortium for Waldenström´s Macroglobulinemia (Dogliotti et al. 2023)
|
|
IgM monoclonal protein |
Bone marrow infiltration |
Symptoms attributable to IgM |
Symptoms due to tumor infiltration |
|
Symptomatic WM |
+ |
+ |
+ |
+ |
|
Asymptomatic WM |
+ |
+ |
- |
- |
|
IgM-related disorders |
+ |
- |
+ |
- |
|
IgM-MGUS |
+ |
- |
- |
- |
Waldenström macroglobulinemia: Diagnostic methods
In the diagnostics of the various lymphoma entities, cytomorphology and histology guide downstream diagnostics. On the one hand, the assessment of the blood and bone marrow smear provides an initial indication as to whether leukemic presentation is present or possible. Cytomorphology and histology are also useful for assessing the degree of lymphoma maturation.
Immunophenotyping allows a clear determination of lineage affiliation to the T- or B-lineage in lymphomas. Furthermore, multiparametric flow cytometry is often indispensable for the differentiation between different lymphoma entities.
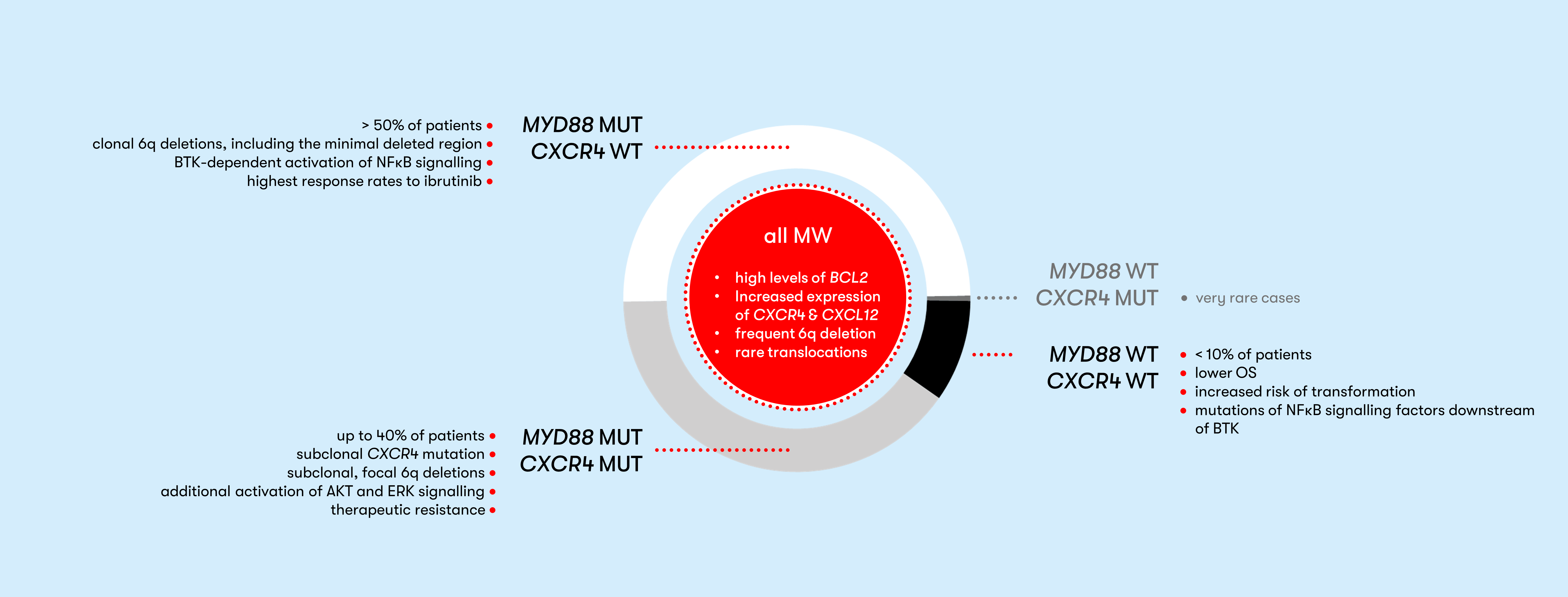
The neoplastic cells express pan-B cell markers (CD19, CD20, CD79, CD22), although CD22 is usually weakly expressed. The lymphoma cells are usually positive for the markers CD25, CD27, FMC7, CD52, CD38 and express IgM. Typically, the degenerated cells are negative for CD5, CD10, CD103, and CD23. BCL2 is expressed in the vast majority of cases (98%) and is a potential therapeutic target (Paiva et al. 2014, Swerdlow et al. 2017, Dimopoulos & Kastritis 2019, WHO 2022).
The most common chromosomal alteration in Waldenström macroglobulinemia is a deletion in the long arm of chromosome 6 (del(6q)). The minimally deleted region includes several genes important for pathogenesis and pathobiology, these are regulatory factors of NFкB signaling (TNFAIP3, HIVEP2), apoptosis (FOXO3), and plasma cell differentiation (PRDM1), as well as the BTK regulator IBTK, the BCL2 regulator BCLAF1, and the ARID1B gene (Guerrera et al. 2018, Treon et al. 2020). The del(6q) occurs in approximately half of all patients (Braggio et al. 2012), but is not specific for Waldenström macroglobulinemia.
A gain of the short arm of chromosome 6 (6p gain) is found as the second most frequent chromosomal aberration. It can result, for example, from the formation of an isochromosome 6p (Braggio et al. 2009). However, the 6p gain occurs exclusively in combination with a 6q deletion. Characteristic for Waldenström macroglobulinemia seem to be whole or partial gains of chromosome 4 as well as a gain of the long arm of chromosome 8. In addition, interstitial deletions occur in the long arm of chromosome 13, affecting the same minimally deleted region as in CLL. Patients with Waldenström macroglobulinemia may have gains of chromosomes 3 and 18, which have also been described in patients with MZL and CLL (Braggio et al. 2009, Braggio et al. 2012).
Fluorescence in situ hybridization can be used to investigate the most common chromosomal alterations in Waldenström's macroglobulinemia (gains of chromosomes 3, 4, and 18, deletion in the long arm of chromosome 6, gain of the long arm of chromosome 8, deletions in the long arm of chromosome 13; see also section "Chromosome analysis"). While rearrangements of the IGH locus are very rare in Waldenström macroglobulinemia compared to other mature B-cell neoplasms (Braggio et al. 2012), ATM deletions (del(11q)) are detectable in 7% of patients and TP53 deletions (del(17p)) in 8% of patients and can also be detected by FISH analysis. Deletion of the TP53 gene is associated with a rather unfavorable outcome (Nguyen-Khac et al. 2013).
Molecularly, the L265P mutation in the MYD88 gene is characteristic, but not specific, for Waldenström macroglobulinemia. This mutation occurs in about 90% of all lymphoplasmacytic lymphomas. It has also been found in patients with IgM MGUS at a lower frequency than in Waldenström macroglobulinemia (Treon et al. 2012, Varettoni et al. 2013). MYD88 mutations in IgM-MGUS represent an independent risk factor for disease progression (Varettoni et al. 2013). In Waldenström macroglobulinemia, the presence of a mutation in the MYD88 gene is associated with better overall survival compared with MYD88 wild-type (Treon et al. 2014, Treon et al. 2018).
In addition to the characteristic MYD88 L265P mutation, the nonsense S338X mutation in the CXCR4 gene is present in approximately 30% of patients with Waldenström macroglobulinemia (Hunter et al. 2014). Other nonsense and frameshift mutations are known (Poulain et al. 2016). In other mature B-cell neoplasms, these mutations occur in less than 10% of patients. Thus, the CXCR4 mutation is relatively specific for Waldenström macroglobulinemia (Treon et al. 2020). A CXCR4 mutation and clonal del(6q) are mutually exclusive; instead, focal subclonal deletions of pathogenetically relevant genes localized on chromosome 6q are found (Fig. 1) (Guerrera et al. 2018).
CXCR4 mutations occur almost exclusively together with a MYD88 mutation (Hunter et al. 2018, WHO 2022). The MYD88 WT (wild type) and CXCR4 MUT (mutation) genotype has been described but occurs very rarely (Hunter et al. 2018, Kaiser et al. 2021).
Waldenström macroglobulinemia: Prognosis
A risk assessment for prognosis can be made with the two scores „International scoring system for Waldenström’s macroglobulinemia“ (ISSWM) (Morel et al. 2009) and „revised international prognostic score system for Waldenström’s macroglobulinemia“ (rIPSSWM) (Kastritis et al. 2019).
Prognostically relevant gene mutations or chromosomal aberrations are not considered in either score.
Waldenström macroglobulinemia: Therapy
Up to 30% of patients have no symptoms at diagnosis. This group is classified as asymptomatic or smoldering Waldenström macroglobulinemia (SMW) (Pophali et al. 2019). The rate of progression to symptomatic Waldenström macroglobulinemia is 12% per year (Bustoros et al. 2019). For these patients, there is a need for close monitoring. The risk for progression to symptomatic Waldenström macroglobulinemia can be determined using an online tool. Relevant to treatment response are CXCR4 mutations, which can negatively affect response to BTK inhibitors in MYD88 L265P mutated patients (Treon et al. 2015, Treon et al. 2018, Tam et al. 2020). In symptomatic patients, genetic characterization plays an increasingly important role in therapy selection. An overview of therapy algorithms can be found in the Onkopedia guideline Waldenström macroglobulinemia (lymphoplasmacytic lymphoma) and in a clinical update by M. Gertz (Gertz 2023). In addition, an overview of therapy management with BTK inhibitors is provided by Buske et al. (Buske et al. 2023).
Status: August 2023