Unser erster Whole Genome Sequencing Befund
Die Ganzgenom-Sequenzierung in der Routinediagnostik
Ein eigenes auf Ganzgenom-Sequenzierung ausgelegtes Befund-Layout
Im Vordergrund des neuen Layouts steht hier die übersichtliche
Darstellung der Ergebnisse im Befund, ohne das Ausmaß der analysierten
Daten zu schmälern. In unserer Standard-Auswerte-Pipeline werden
Punktmutationen (SNV, single nucleotide variants), strukturelle
Varianten (SV), Kopienzahlveränderungen (CNV, copy number variations)
und Verluste der Heterozygotie (CN-LOH, copy-neutral loss of
heterozygosity) untersucht und berichtet. Neben einer kurzen Darstellung
der gefundenen Veränderungen enthält der Befund eine grafische
Aufbereitung sämtlicher Ergebnisse als Circos-Plot (Abbildung).
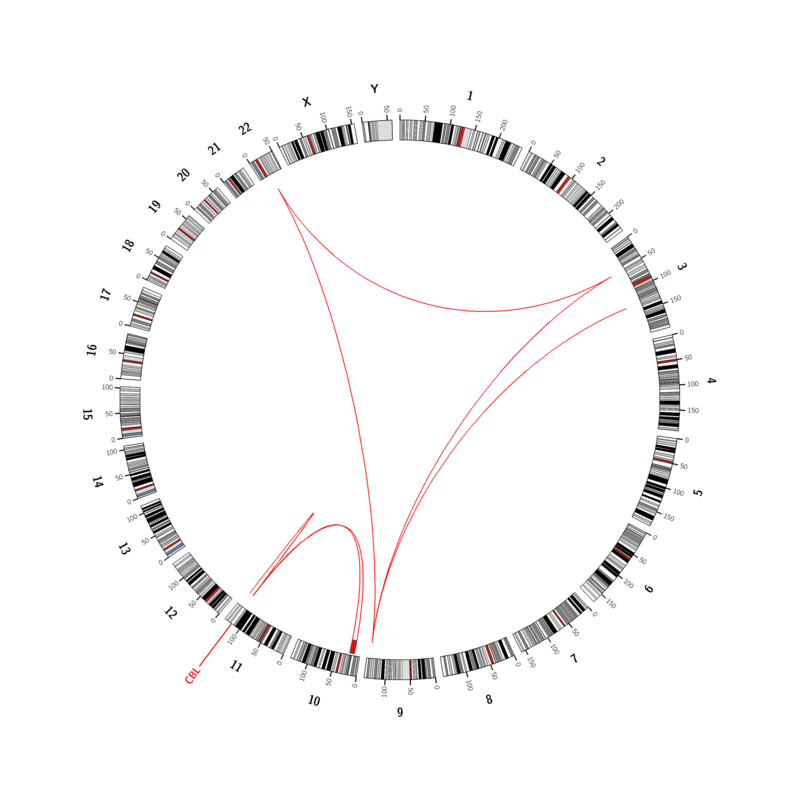
Bei unserem ersten klinischen Befund einer Ganzgenom-Sequenzierung handelte es sich um eine CML in Blastenkrise, die neben einer komplexen 3-Wege Translokation zwischen Chromosom 3, 9 und 22, die zum BCR-ABL1 Fusionstranskript führte, eine zweite dazugewonnene KMT2A-MLLT10 Translokation zwischen Chromosom 10 und 11 offenbarte. Darüber hinaus konnten eine Mutation (SNV) in CBL sowie der Verlust von chromosomalem Material von Chromosom 10 identifiziert werden.
In unserem Circos-Plot werden die Chromosomen kreisförmig gelistet, Translokationen durch rote Verbindungslinien dargestellt, Zugewinne (blau) und Verluste (rot) im Inneren des Chromosomenkreises gekennzeichnet und das Gen mit einer Mutation an seiner Position im Genom genannt. Diese grafische Darstellung der Ergebnisse soll dem behandelnden Arzt einen schnellen Überblick über die genomweiten Veränderungen geben. Natürlich enthält dieser Befund wie gewohnt auch die relevanten Veränderungen in Textform, Tabellen mit Detailinformationen und die daraus resultierende Interpretation in Bezug auf Diagnose und Prognose.
Der erste klinische Befund wurde versendet
Wir freuen uns, dass wir Mitte Juni den ersten klinischen Befund zur Ganzgenom-Sequenzierung im neuen, um eine Grafik erweiterten Layout, versenden konnten. Die Zusatzinformation, die eine Ganzgenom-Sequenzierung bringen kann, wird sich in der Zukunft manifestieren. Umso schöner ist es, dass wir bereits jetzt für komplexe Fälle, aber auch Erkrankungen wie das multiple Myelom oder die akute lymphatische Leukämie (ALL) durch die Ganzgenom-Sequenzierung und Transkriptomanalysen zur Weiterentwicklung der diagnostischen Methoden beitragen, therapie-steuernde Befunde erheben und die Ergebnisse krankheitsrelevant übersichtlich darstellen können.
Die Autorin

»Sie haben Fragen zum Artikel oder wünschen weitere Informationen? Schreiben Sie mir gerne eine E-Mail.«
Dr. rer. nat. Manja Meggendorfer, MBA
Diplombiologin
Leitung des Bereichs Molekulargenetik
Leitung Forschung und Entwicklung
manja.meggendorfer@mll.com