62. ASH Annual Meeting & Exposition – ein Nachbericht
Auch im Jahr 2020, diesmal als virtueller Kongress, war das MLL beim 62. Annual Meeting & Exposition der American Society of Hematology (ASH) gut durch eigene Beiträge und in Kooperationen vertreten. Insgesamt hat das MLL mit seinem Team vier Vorträge gehalten und 11 Poster inklusive Kurzvortrag vorgestellt.
Die Schwerpunkte dabei waren die genauere Charakterisierung und Klassifizierung von Leukämien und Lymphomen ebenso wie die Anwendung neuer Methoden bis hin zu Genom-Sequenzierung und künstlicher Intelligenz, sowohl in Forschung als auch in der Routinediagnostik.
Insgesamt 12 Wissenschaftler aus dem MLL haben die Ergebnisse der Forschungsprojekte präsentieren dürfen. Als Vortrag aus dem Bereich der Zytomorphologie hat Herr Dr. C. Pohlkamp die aktuellen Ergebnisse vorgetragen, mit Hilfe von künstlicher Intelligenz (Kooperationen mit AWS) das periphere Blutbild zu differenzieren. Die dabei erreichten richtigen Voraussagen zur einzelnen Zellsorte benigner und maligner Zellen im peripheren Blut liegt mit dem getesteten Algorithmus bei 92%. Die dabei entwickelten Tools können Cloud basiert angewendet werden und befinden sich gerade in der prospektiven Testung (BELUGA-Studie) parallel zum Goldstandard der peripheren Blutbilddifferenzierung durch MTAs und Hämatologen.
Für die klassische Chromosomenanalyse hat Frau Prof. C. Haferlach den aktuellen Stand der Dinge vorgestellt: Dabei ist es dem MLL gelungen, zusammen mit der Firma MetaSystems Algorithmen zu entwickeln, die bei der Karyotypisierung das Fachpersonal von MTAs und Biologen unterstützen können. Schon weit über 20.000 Patienten sind damit mit Unterstützung von Algorithmen der künstlichen Intelligenz in der Routinediagnostik prozessiert worden und führen zu einer deutlichen Verkürzung der Befundungszeit der Chromosomenanalyse zum Teil um zwei Tage im Vergleich zur vorherigen Dauer des Workflows.
In der Molekulargenetik spielt zunehmend die Panel-Sequenzierung mit Next-Generation-Sequencing (NGS) eine Rolle. Speziell auch bei unklaren Fragestellungen und zum Ausschluss von MDS ergeben sich hier deutliche Vorteile für die klinische Entscheidungsfindung und für weitergehende Diagnostik und Therapiewahl. Frau Dr. C. Bär hat hier die Ergebnisse vorgestellt und konnte zeigen, dass bei unklaren Konstellationen wie Zytopenien die Analyse eines Panels mit bei myeloischen Neoplasien häufig mutierten Gene zu einer deutlichen Verbesserung der klinischen diagnostischen Algorithmen und zu einer Einschränkung der differenzialdiagnostisch in Betracht kommenden Diagnosen führt. Für den Patienten und für den Hämatologen ergibt sich dadurch eine kürzere Zeit bis zur definitiven Diagnose oder Ausschlussdiagnose, die sonst üblichen vielfachen Wiederholungs-Punktionen bei gleicher klinischer Konstellation des Blutbildes können reduziert werden.
Darüber hinaus hat das MLL zusammen mit dem ASH in den letzten zwei Jahren ein Projekt gesteuert, dessen Ergebnis Herr N. Nadarajah beim ASH-Kongress vorgetragen hat. Dabei geht es um die Harmonisierung von molekulargenetischen Befunden, die unter Leitung des MLL und durch gemeinsame Datenauswertung von sechs unterschiedlichen Laboren in Europa und in den USA zu einer gemeinsamen konsentierten Beurteilung von Varianten in Genen geführt hat, die bei hämatologischen Erkrankungen verändert sind. Gemeinsam wurden dabei auch die Algorithmen zur Auswertung und der Laborworkflow zwischen den Laboren diskutiert und abgeglichen. Das Ergebnis wurde im Oktober 2020 zur Verwendung bei der Befundung von hämatologischen Neoplasien auf der ASH Homepage zur Verfügung gestellt und wird jetzt von uns weiter bearbeitet und erweitert.
Ziel der wissenschaftlichen Aktivität des MLL wird es also weiterhin sein, die Diagnostik für unsere Patienten zu verbessern, die Daten im Rahmen von wissenschaftlichen Publikationen und Kooperationen zu teilen und zur Verfügung zu stellen und insbesondere neue technische Möglichkeiten wie das NGS aber eben auch die Anwendung von künstlicher Intelligenz zur Unterstützung der Routinediagnostik im Labor voranzutreiben.
Im Folgenden haben wir alle ASH-Projekte mit MLL-Beteiligung zusammengefasst:
Autoren: C. Baer, A. Stengel, W. Kern, C. Haferlach, T. Haferlach
Die Molekulargenetik spielt eine zunehmende Schlüsselrolle bei Patienten mit Zytopenien und dem MDS. Daher nutzen wir im MLL seit einigen Jahren die Panel-Sequenzierung im breiten Routineeinsatz. Mit diesen Daten haben wir eine systematische Auswertung von 576 morphologisch und immunphänotypisch gut charakterisierten Fällen und deren Mutationsprofiel durchgeführt. Es wurden 213 Patienten als MDS nach WHO Kriterien eingestuft; davon wiesen 85% mindestens eine Mutation in der Panel-Sequenzierung auf. Auch bei einem Drittel der Fälle ohne klassische MDS Diagnose waren bereits Mutationen zu finden. Folglich sind diese als klonale Zytopenie unbestimmter Signifikanz (CCUS) zu bezeichnen.
Ziel der Studie war eine weitere Subklassifikation der CCUS Patienten. Ein Viertel zeigte ausschließlich sogenannte DTA-Mutationen (Mutationen in den Genen DNMT3A, TET2 oder ASXL1, die häufig im Alter auftreten). Allerdings ließen sich in weiteren 20% der CCUS Kohorte Mutationen in Genen nachweisen, die mit einem hohen Risiko einhergehen, eine myeloische Erkrankung zu entwickeln (z.B. RUNX1 oder SF3B1). Dabei haben wir ein besonderes Augenmerk auf die Patienten mit SF3B1 Mutationen gelegt, da die internationale Arbeitsgruppe (IWG-PM) um L. Malcovati empfiehlt, diese Mutationen unabhängig von der Morphologie als Kriterium für die MDS Diagnose heranzuziehen (Malcovati et al. Blood, 2020). Auch wir finden bereits bei 5% der CCUS Patienten eine SF3B1 Mutation.
Ähnlich wie diese SF3B1 Mutationen könnten auch weitere Gene in der Zukunft nicht nur eine Rolle in der Prognoseabschätzung sondern auch der Diagnosestellung des MDS spielen und die Molekulargenetik damit weiter als feste Säule in der MDS Diagnostik verankern.
Alle Informationen sowie den Abstract zur Studie finden Sie hier.
Autoren: C. Baer*, S. Kimura*, I. Iacobucci, D. J. Feith, W. Walter, M. Meggendorfer, T. L. Olson, A. Ratan, M.-L. Mueller, M. Rana, C. Qu, R. Kalathur, C. Haferlach, W. Kern, T. P. Loughran, Jr., C. G Mullighan, T. Haferlach (*Contributed equally)
Die chronische lymphoproliferative Störung der natürlichen Killerzellen (CLPD-NK) ist eine seltene hämatologische Erkrankung, bei der die Unterscheidung zwischen neoplastischem und reaktivem Zustand die größte diagnostische Herausforderung ist.
Im Rahmen unserer Ganzgenomsequenzierungen konnten wir bislang nicht beschriebene Mutationen im Chemokin CCL22 finden, die bei circa einem Drittel aller Patienten vorkommen. Zusammen mit dem Team von Dr. Mullighan (St. Jude Children’s Research Hospital, Memphis) und Dr. Loughran (University of Virginia School of Medicine, Charlottesville) konnten wir diese Mutationen in einer unabhängigen Kohorte validieren und eine Reihe funktioneller Untersuchungen durchführen. Werden beispielsweise Zellen mit der Mutation in die Maus transplantiert, wachsen diese schneller an als die Kontrollen. Außerdem hat die Mutation einen Einfluss auf die Migration (Chemotaxis) der Zellen in vitro.
Die Entdeckung der CLL22 Mutation erlaubt uns, die Pathogenese der CLPD-NK besser zu verstehen, und es steht uns nun neben STAT3 ein weiterer molekulargenetischer Marker zum Klonalitätsnachweis bei Auffälligkeiten der NK-Zellen zur Verfügung.
Weitere Informationen sowie den Abstract zur Studie können Sie hier nachlesen.
Autoren: C. Haferlach, S. Hänselmann, W. Walter, S. Volkert, M. Zenger, W. Kern, A. Stengel, T. Lörch, T. Haferlach
Die Chromosomenanalyse spielt bei zahlreichen hämatologischen Neoplasien eine wichtige Rolle bei der Klassifikation sowie bei der Einschätzung der Prognose.
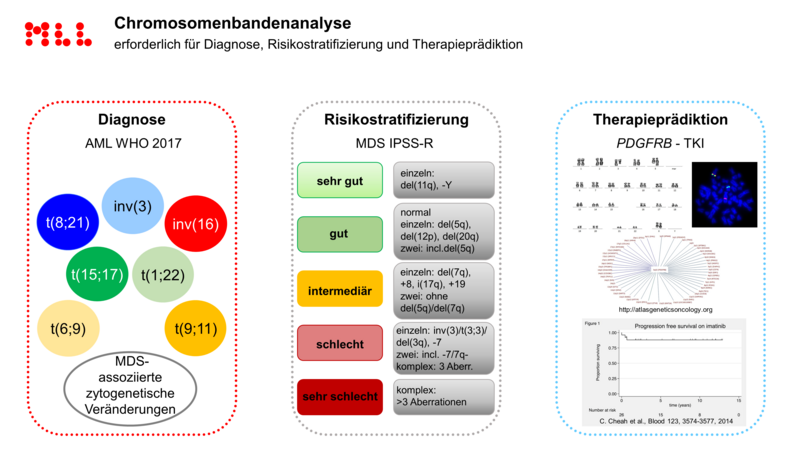
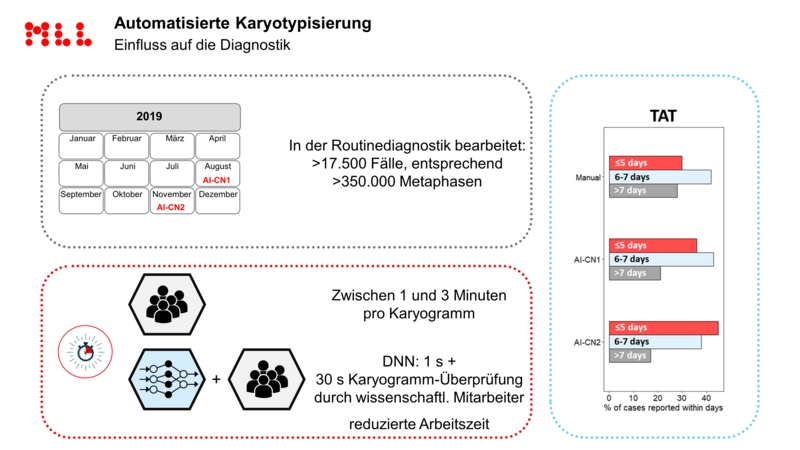
Diese Methode ist in Bezug auf die Labormethoden sowie die Auswertung sehr zeitaufwendig und die Qualität der Befunde abhängig von der Erfahrung des Zytogenetikers. Kurze Befundungszeiten werden immer wichtiger, da zunehmend Therapie-Entscheidungen durch den Karyotyp beeinflusst werden. In den letzten Jahren wurden die Laborprozesse automatisiert – jetzt war das Ziel auch die Auswertung zu automatisieren unter Nutzung der Fortschritte im Bereich künstlicher Intelligenz (KI). Auf der Basis von 100.000 Karyogrammen mit normalem Karyotyp aus unserem digitalen Archiv wurde ein Chromosomen-Klassifikator trainiert. Eine prospektive Validierung mit 500 Karyogrammen ergab eine korrekte Zuordnung von 22.675 der 23.000 Chromosomen (98.6%). Dieser Klassifikator wurde im August 2019 in die Routine eingeführt. Seitdem wurden >350.000 Metaphasen mit Unterstützung des Klassifikators ausgewertet. Dieses führte
1) zu einer Reduktion der Arbeitszeit: ein erfahrener Zytogenetiker benötigt in Abhängigkeit von der Chromosomenqualität zwischen 1 und 3 Minuten für die Erstellung eines Karyogramms, während der KI-basierte Klassifikator lediglich 1 Sekunde benötigt und der Zytogenetiker ca. 30 Sekunden für die Validierung.
Autoren: K. Loy, M. Zenger, M. Meggendorfer, S. Hutter, W. Kern, T. Haferlach, C. Haferlach, C. Baer
Die Chronische Myeloische Leukämie (CML) ist dank der Entdeckung von spezifischen Tyrosinkinaseinhibitoren in der chronischen Phase eine sehr gut behandelbare Erkrankung. Dahingegen haben Patienten, die in die akzelerierte Phase oder in Blastenkrise übergehen eine sehr schlechte Prognose. Um einen detaillierten Einblick in die Progression individueller Patienten bei der CML zu bekommen, wurden 11 Patienten bei Diagnose (D), in Remission (REM) und in Blastenkrise (BK) mittels Whole Genome Sequencing (WGS) analysiert. Es wurden drei unterschiedliche Mechanismen der Progression identifiziert: Zusätzliche chromosomale Veränderungen neben der BCR-ABL1 Translokation wurden bei 91% der Patienten nachgewiesen, Mutationen in Genen, die bekanntermaßen bei hämatologischen Neoplasien alteriert sein können, bei 64% und resistenzvermittelnde Mutationen im ABL1 des BCR-ABL1 Fusionstranskripts bei 55%. Alle diese Veränderungen konnten ausschließlich in der Blastenkrise nachgewiesen werden. Ein früher Nachweis von zusätzlichen Veränderungen könnte daher zu einer früheren therapeutischen Intervention und zu einer besseren Prognose beitragen.
Autoren: A. Maierhofer, C. Baer, C. Pohlkamp, M. Meggendorfer, W. Kern, C. Haferlach, T. Haferlach
Das Screening einer Kohorte von 1.228 Patienten mit sporadischer AML oder MDS ergab, dass 8,3% der Patienten eine potentielle Keimbahnvariante (definiert als Variante mit Allelfrequenz >0,3) in den Prädispositionsgenen DDX41, ETV6 oder GATA2 aufwiesen. Dies stützt die Hypothese, dass Keimbahnmutationen in Prädispositionsgenen für myeloische Neoplasien häufiger vorkommen könnten als angenommen und hebt die Bedeutung einer systematischen Testung auf diese Varianten, unabhängig von der Familienanamnese oder dem Alter des Patienten, hervor. 38 der 39 Patienten, die eine potentielle Keimbahnvariante in ETV6 oder GATA2 trugen, wiesen mindestens eine weitere genetische Variante in zusätzlichen bekannten AML- oder MDS-assoziierten Genen auf. Bei 25% der 64 Patienten, die eine potentielle Keimbahnvariante in DDX41 (und bei 12 Fällen eine zusätzliche somatische DDX41-Mutation) trugen, konnten dagegen keine weiteren genetischen Varianten in anderen Genen nachgewiesen werden. Dies weist darauf hin, dass DDX41 und insbesondere der Zugewinn einer zweiten somatischen Mutation die Entwicklung einer Leukämie vorantreiben, wohingegen bei ETV6 und GATA2 zusätzliche Mutationen erforderlich zu sein scheinen.
Autoren: M. Meggendorfer, W. Walter, N. Nadarajah, C. Haferlach, W. Kern, T. Haferlach
Mit zunehmenden Sequenzierkapazitäten wird auch das Spektrum der untersuchten Gene breiter. Ziel unserer Studie war es daher, den tatsächlichen Gewinn an genetischer Information mit zunehmender NGS-Panelgröße bei MDS Patienten zu bestimmen, beginnend mit kleinen Panels der diagnostischen Richtlinien bis hin zur kompletten kodierenden Region - dem Exom. Die Untersuchung der 12 NCCN-Panel-Gene ergab, dass 81% der MDS Patienten mindestens eine Mutation tragen, mit Ausweitung der Analyse auf das McClure-Panel (34 Gene) stieg diese Zahl auf 84% und weiter auf 87% und 100 %, wenn die Cosmic Cancer Genes (723 Gene) bzw. das Exom berücksichtigt wurden. Bei 588 Patienten wurden also insgesamt 1.058 Mutationen in nur 12 Genen detektiert. Die Anzahl an Mutationen stieg auf 4.000 durch die Untersuchung des Exoms. Im Gegenteil dazu stieg jedoch die Zahl an Varianten unklarer Signifikanz von nur 50 auf über 21.000 bei Untersuchung des Exoms. Damit wird deutlich, dass MDS-Patienten mit einem kleinen diagnostischen Genpanel untersucht werden können und durch steigende Zahlen an Genen meist Varianten unklarer Signifikanz zusätzlich detektiert werden, deren klinische Relevanz noch durch weitere Studien bestimmt werden muss.
Alle Informationen zu diesem Projekt finden Sie auf der Webseite der ASH.
Autoren: L. Montefiori, S. Seliger, Z. Gz, X. Ma, B. Xu, X. Chen, K. Dickerson, T. Westover, A. Stengel, I. Iacobucci, S. Kimura, C. Qu, J. Ma, M. Valentine, W. Kern, T. Haferlach, G. Wu, J. Klco, C. Haferlach, C.G. Mullighan
Akute Leukämien mit gemischtem Phänotyp sind nach wie vor schwierig zu diagnostizieren und über ihren biologischen Ursprung ist wenig bekannt. In Zusammenarbeit mit dem St. Jude Children’s Research Hospital in Memphis, Tennessee, wurden über 2500 Proben von Patienten mit akuter Leukämie mit Whole Transcriptome Sequencing, sowie ein Teil der Patienten mit Whole Genome Sequencing, analysiert. Es konnte ein Subtyp von akuten Leukämien mit T-Zell- und myeloischen Markern identifiziert werden, die über ihr transkriptomisches Profil und ihre monoallelische Expression des hämatopoetischen Transkriptionsfaktors BCL11B charakterisiert sind. 80% der Fälle zeigten Veränderungen von FLT3 und in allen Fällen wurden strukturelle Veränderungen von BCL11B nachgewiesen. Die meisten Patienten zeigten Translokationen von BCL11B mit ARID1B, CCDC26, CDK6, ETV6, RUNX1 oder ZEB2, welche für diese Subgruppe spezifisch waren und in keiner anderen Neoplasien beobachtet wurden. Diese Loci enthalten Enhancer, die in frühen hämatopoetischen Vorläuferzellen aktiv sind. In den anderen Fällen wurde eine Amplifikation einer 2,5 kb großen Region distal von BCL11B nachgewiesen, die zur Entstehung eines neuen Enhancers führt – ein Mechanismus, der so zuvor noch nicht beschrieben wurde. Die Ergebnisse dieser Arbeit zeigen somit Enhancer-Hijacking eines ansonsten linienspezifischen Transkriptionsfaktor-Gens als definierendes Merkmal eines Subtyps von akuten Leukämien mit unklarer Abstammung. Es konnte zudem gezeigt werden, dass zumindest in einem Teil der Patienten eine hämatopoetische Stammzelle Ursprung der Leukämie ist.
Autoren: J. Müller, C. Haferlach, H. Ruge, H. Müller, I. Fuhrmann, M. Meggendorfer, M.-L. Müller, W. Kern, T. Haferlach, A. Stengel
Die akute lymphoblastische T-Zell-Leukämie (T-ALL) ist eine seltene aggressive Neoplasie, die ca. 20 % aller ALL-Fälle ausmacht. Sie tritt häufiger bei Erwachsenen auf als bei Kindern, obwohl die Inzidenz mit zunehmendem Alter abnimmt. Die Subklassifikation der T-ALLs nach WHO basiert bisher ausschließlich auf dem Immunphänotyp. Obwohl eine Reihe von häufigen molekularen Aberrationen bei T-ALL beschrieben wurden, fehlt bisher eine detaillierte molekulare Klassifikation.
Durch die Kombination von Whole Genome Sequencing (WGS) und Whole Transcriptome Sequencing (WTS) konnten wir 114 T-ALL-Fälle auf molekularer Ebene in sechs verschiedene genetische Subgruppen einteilen. Circa die Hälfte der Fälle ließen sich in die bereits vier bekannten Subgruppen einteilen, die durch die Überexpression der Onkogene TLX1, TLX3, HOXA oder TAL1 (Gruppe 1-4) definiert sind. Die übrigen wurden basierend auf die onkogene Aktivierung des NOTCH1-Signalwegs in zwei weitere Subgruppen (Gruppe 5+6) eingeteilt. Dabei zeigte sich, dass nur in den letzten beiden Gruppen (5+6) Mutationen in den Genen DNMT3A, TET2 und ASXL1 auftraten.
Insbesondere DNMT3A Mutationen wurden signifikant häufiger bei älteren Patienten nachgewiesen und sind mit einem schlechteren Gesamtüberleben assoziiert. WGS Analysen identifizierten außerdem einen bisher unbekannten Typ von NOTCH1-Mutationen, die als NOTCH1-ITD bezeichnet wurden. Dabei handelt es sich um ein Duplikationsevent in der NOTCH1-Domäne.
Diese Daten bieten eine Grundlage, um T-ALLs auf molekularer Ebene weiter zu klassifizieren, prognostische und MRD-Marker zu identifizieren und vor allem, um das Wissen in Zukunft in effiziente zielgerichtete Therapien umzusetzen.
Weitere Informationen zu dieser Publikation können Sie hier nachlesen.
Autoren: M-L. Müller, N. Nadarajah, K. Jhalani, I. Heo, W. Wetton, C. Haferlach, T. Haferlach, W. Kern
Der Einsatz von künstlicher Intelligenz im Zusammenhang mit durchflusszytometrischen Daten bietet sehr viel Potenzial für eine weitreichende Automatisierung der Datenauswertung. Bislang wurde dies hauptsächlich mit Hilfe von Zwischenschritten über diverse Visualisierungstechniken erreicht, die aber mit einem Datenverlust durch Dimensionsreduktion einhergehen können. Wir konnten hier eine neue Herangehensweise entwickeln, welche auf der Berechnung von unveränderten Rohdaten basiert. Am Beispiel von neun Klassen von B-Zelllymphomen wurden rund 6400 entsprechende Proben analysiert. Jede davon wurde mit 33 Parametern durchflusszytometrisch gemessen und anschließend sowohl manuell als auch mit Hilfe von künstlicher Intelligenz ausgewertet. In Zusammenarbeit mit unserem Kollaborationspartner Amazon Web Services (AWS) wurden verschiedene Algorithmen entwickelt und getestet: ein Decision Tree Modell, ein Deep Learning Modell und ein XG Boost Modell. Unter Berücksichtigung einer Vorhersagewahrscheinlichkeit von > 95% und Klongrößen der pathologischen Entitäten von > 0,1% erreichte der XG Boost Algorithmus die höchste Richtigkeit (93%) einhergehend mit geringstmöglichem Anteil nicht zugeordneter Fälle (28%). Der XG Boost Algorithmus muss noch weiterentwickelt werden, um auch komplexere Lymphomkonstellationen zu erkennen, sowie andere pathologische Entitäten einschließen zu können.
Autoren: N. Naradajah, A. Wagner, R. Bejar, M. Ewalt, A.S. Kim, M.M. Le Beau, X. Ma, M. Meggendorfer, J.M. Shammo, G. Ryan, A.J. Siddon, D.P. Steensma, M.J. Walter, A. Zehir, J. Zhang, T. Haferlach
Next-Generation-Sequencing (NGS) nimmt zunehmend eine wichtigere Rolle in der Diagnostik ein. Obwohl die Technologie in der letzten Dekade rasante Fortschritte gemacht hat, bleibt die klinische Interpretation der Daten und insbesondere die Klassifikation von somatischen Varianten eine schwierige Aufgabe. Um der Hämatologie Gemeinschaft bei diesem Problem zu helfen, hat sich eine Arbeitsgruppe innerhalb der ASH (American Society of Hematology) unter Federführung von Prof. Dr. Dr. Haferlach gegründet und eine zuverlässige und allgemein verfügbare Ressource entwickelt, mit dem Ziel die NGS Diagnostik konsistenter und robuster zu machen und damit die Patientenversorgung zu verbessern. Das Ergebnis der Arbeit der interdisziplinären Gruppe, bestehend aus Hämatologen, Molekularbiologen und Bioinformatikern, ist als interaktive Webapplikation über die ASH Homepage zugänglich. Die Klassifikationen für 202 Varianten mit hoher Konfidenz wurden zusammengefasst.
Das Original-Paper sowie den Abstract zu diesem Projekt finden Sie auf der ASH-Webseite.
Autoren: C. Pohlkamp, K. Jhalani, N. Nadarajah, I. Heo, W. Wetton, R. Drescher, S. Hänselmann, T. Lörch, C. Haferlach, W. Kern, T. Haferlach
Im Bereich der Zytomorphologie präsentierte das MLL einen machine learning-basierten Klassifikator für Blutzellen, der in Zusammenarbeit mit den Firmen AWS und MetaSystems entwickelt wurde. Zunächst erfolgt der vollautomatische Scan von Ausstrichen bzw. Zellen des peripheren Bluts. Im Anschluss ist der Klassifikator in der Lage, mit hoher Genauigkeit sämtliche physiologische und pathologische Zelltypen des peripheren Blutes in Form digitaler Bilder zu erkennen und zu unterscheiden. Dies gilt insbesondere auch für seltene pathologische Zelltypen wie Haarzellen und atypische Promyelozyten der APL. Das Tool kann perspektivisch selbst erfahrene Hämatologen in der zytomorphologischen Diagnostik unterstützen und unterliegt nicht den typischen Limitationen eines menschlichen Untersuchers (Konzentration, Ermüdung, untersuchte Zellzahl...). Ein weiterer angedachter Schritt ist die Entwicklung eines Klassifikators für Knochenmarkzellen.
Alle Informationen zu diesem Projekt können Sie hier nachlesen.
Autoren: A. Stengel, M. Meggendorfer, W. Kern, T. Haferlach, C. Haferlach
Normalerweise werden DNA-Schäden durch verschiedene DNA-Reparatur-Wege behoben. Durch Störungen dieses Gleichgewichts akkumulieren somatische Zellen jedoch Mutationen, was als eine der Hauptursachen nicht nur für das Altern, sondern auch für die Entstehung von Krebs angesehen wird. In diesem Projekt haben wir nach Korrelationen der Mutationshäufigkeiten (mittels Whole Genome Sequencing) von 122 Genen bei 2656 Patienten mit 11 verschiedenen hämatologischen Neoplasien mit dem Alter der jeweiligen Patienten gesucht. Insgesamt wurden 5709 Mutationen in den 122 analysierten Genen nachgewiesen. Es wurden eine Reihe von Mutationen gefunden, die über alle Entitäten hinweg mit einem höheren Alter der Patienten korreliert waren, darunter sogenannte CHIP-Gene (TET2, DNMT3A, ASXL1), aber auch andere Gene (z.B. TP53, EZH2, BCOR, GATA2, IDH2). Eine geringere Anzahl von Mutationen war mit jüngerem Alter korreliert und z.T. mit anderen altersbedingten Aberrationen assoziiert. Einige Mutationen zeigen zudem eine unterschiedliche Alterskorrelation in Abhängigkeit von der Entität (z.B. PHF6-Mutationen), was auf unterschiedliche Mutations- oder Selektionsmechanismen in Abhängigkeit von der jeweiligen Entität schließen lässt.
Alle Informationen sowie den Abstract zur Studie finden Sie hier.
Der Autor

»Sie haben Fragen zum Artikel oder wünschen weitere Informationen? Schreiben Sie mir gerne eine E-Mail.«
Prof. Dr. med. Dr. phil. Torsten Haferlach
Geschäftsführung
Internist, Hämatologe und Onkologe
Stellvertretende Bereichsleitung Zytomorphologie
torsten.haferlach@mll.com